


- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Modern Human Pathology
Volume 1, Issue 2, April 2016, Pages 21–24
Case reportOpen Access
Talc induced pulmonary granulomatosis: An under recognized complication in patients with cystic fibrosis
-
Arlette Habashi-Daniel1,
Michael Crosser2 and
Ossama Tawfik1,*

*Corresponding author: Ossama Tawfik, M.D., Ph.D. Professor, Director of Anatomic and Surgical Pathology; Department of Pathology and Laboratory Medicine; The University of Kansas Medical Center; 3901 Rainbow Boulevard, Kansas City, KS 66160, USA. Tel.: (913) 588-1185; Fax: (913) 588-878; E-mail: otawfik@kumc.edu
Received 4 February 2015 Accepted March 2016 Published 31 March 2016
DOI: http://dx.doi.org/10.14312/2397-6845.2016-4
Copyright: © 2016 Habashi-Daniel A, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
Talc induced pulmonary granulomatosis is a known pulmonary condition that is associated with intravenous drug exposure. Only one documented case of talc induced pulmonary granulomatosis in a patient with cystic fibrosis (CF) has been reported in the literature. We add another case of such condition. Our goal is to increase the awareness of this under recognized potential cause of CF related pulmonary induced fibrosis and hypertension. Patients with risk factors and with findings similar to the reported patient may benefit from further clinical and pathological evaluation for talc induced pulmonary granulomatosis.
Keywords: talc induced pulmonary granulomatosis; talcosis; cystic fibrosis
IntroductionTop
Pulmonary complications secondary to cystic fibrosis (CF) has been well documented and can have multiple etiologies [1, 2]. Recurrent viral, bacterial and/or fungal pulmonary infections, excessive mucus secretion in the lung, impaired mucociliary clearance and progressive bronchiectasis are well known causes that could lead to the exacerbation of CF related lung symptoms [1, 2].
Talc induced pulmonary granulomatosis is a known pulmonary condition that is associated with intravenous (IV) drug exposure [3-5]. Other terms used to describe this condition include talcosis, self-induced granulomatosis, pulmonary granulomatous talcosis and pulmonary angiothrombotic granulomatosis. The distinctive pathologic changes are for the most part due to the fillers used in the manufacture of pills that are intended for oral use [5, 6]. Tablets intended for oral use contain insoluble binding agents "fillers" such as talc (hydrated magnesium silicate), microcrystalline cellulose, potato starch and corn starch. When oral medications are crushed, melted, dissolved in water and IV-injected, fillers in the drug, such are talc particles, become lodged in pulmonary vessels. The talc crystals create a foreign body granulomatous inflammatory reaction with significant damage to the surrounding lung tissue (talc granulomatosis).
We present an unusual case of a 28-year-old female CF patient with declining pulmonary functions that was discovered to be due to the repeated self-IV injection of alprazolam and hydrocodone/APAP via a port-a-cath. The clinical, radiological and histopathological findings with review the literature are discussed.
Case reportTop
28-year-old female with a past medical history of CF, presented to the emergency department with shortness of breath and productive cough of one week. Her past medical history was significant for CF diagnosed at age of 2 months. She had previously had multiple CF exacerbations which have required hospitalization. At age 20 she had an elective port-a-cath placed for central venous access for outpatient antibiotic administration. In 2011, she had sepsis of unclear etiology, leading to port removal which was replaced a year later. In 2012, she developed fungemia (Candida albicans), where her port was found to be infected again and was subsequently removed. Her significant medications during admission were alprazolam and hydrocodone-acetaminophen in addition to her nebulized CF medications. She was a nonsmoker with no history of illicit drugs or alcohol consumption. Her admission physical examination showed that she was a thin Caucasian female in no acute distress. She was afebrile, her oxygen saturation was 97% and her respiratory rate was 20 with a normal blood pressure. Her lungs had bilateral rales in the upper poles which was a chronic finding. She had clubbing of fingers without cyanosis. The remainder of her physical examination was normal. The chest radiograph showed chronic bronchiectasis and fibrosis that was more prominent in the upper lobes, but with no definite infiltrates.
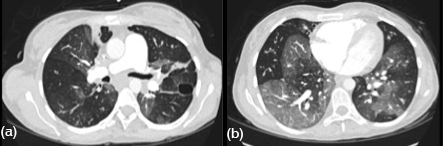
She was admitted with a diagnosis of CF exacerbation. Her hospital course was very complicated requiring multiple intensive care unit admissions for respiratory failure and shock requiring life support interventions. She even had an episode of shock leading to pulseless electric activity and cardiac arrest requiring 6 minutes of cardiopulmonary resuscitation. Her port grew Candida albicans and Veillonella that was treated with IV antibiotics and antifungals. Computed tomography (CT) angiography of the chest was performed revealing bilateral ground glass pulmonary opacities with basilar predominance of unclear etiology (Figure 1). There was a questionable small pulmonary embolus in the right upper lobe. She received anticoagulation but did not improve clinically therefore a pulmonary angiogram was performed which was negative for pulmonary embolism.
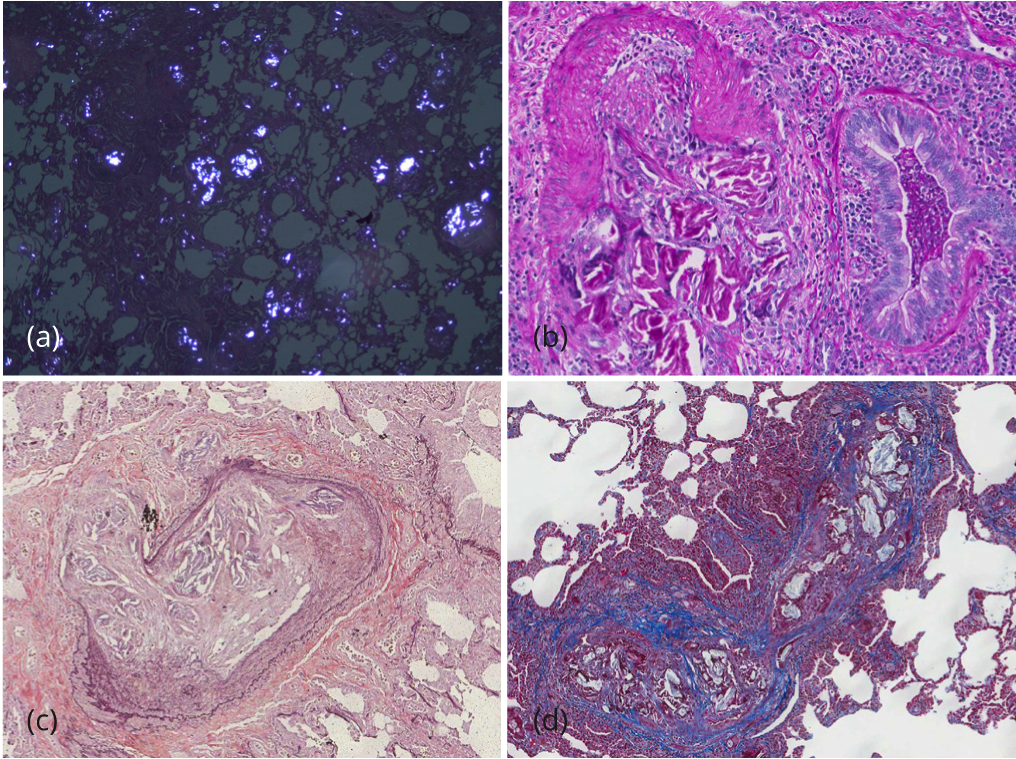
During her prolonged hospitalization, she had numerous echocardiograms of her heart. She had variable findings of right heart strain and pulmonary hypertension. At times, her pulmonary artery systolic pressure (PASP) was greater than 70mmHg with right ventricular dilation and dysfunction. However, within days, her PASP and right ventricular size and function would normalize. Two right heart catheterizations were essentially normal which correlated with normalization of echocardiographic findings. Eventually, thoracic surgery was consulted for video-assisted thoracoscopic lung biopsies. Several wedge biopsies from the left upper and lower lobes were obtained. The pleural surface was unremarkable. Serial sectioning revealed multiple 0.5 to 1.6 cm poorly demarcated tan white discernable lesions. On microscopic evaluation, the slides showed total occlusion of multiple medium and small sized pulmonary vessels with polarizable foreign material admixed with a prominent foreign body giant cell reaction (Figure 2). The lung parenchyma was extensively involved by multiple granulomas with multinucleated giant cells and abundant polarizable material. Permanent histologic examination, with hematoxylin and eosin, PAS, trichrome, and elastic stains, demonstrated the exogenous material had the typical histochemical and structural characteristics of talc crystals, distributed within perivascular connective tissue and vessel walls, with surrounding granulomatous reaction (Talc granulomatosis) (Figure 2).
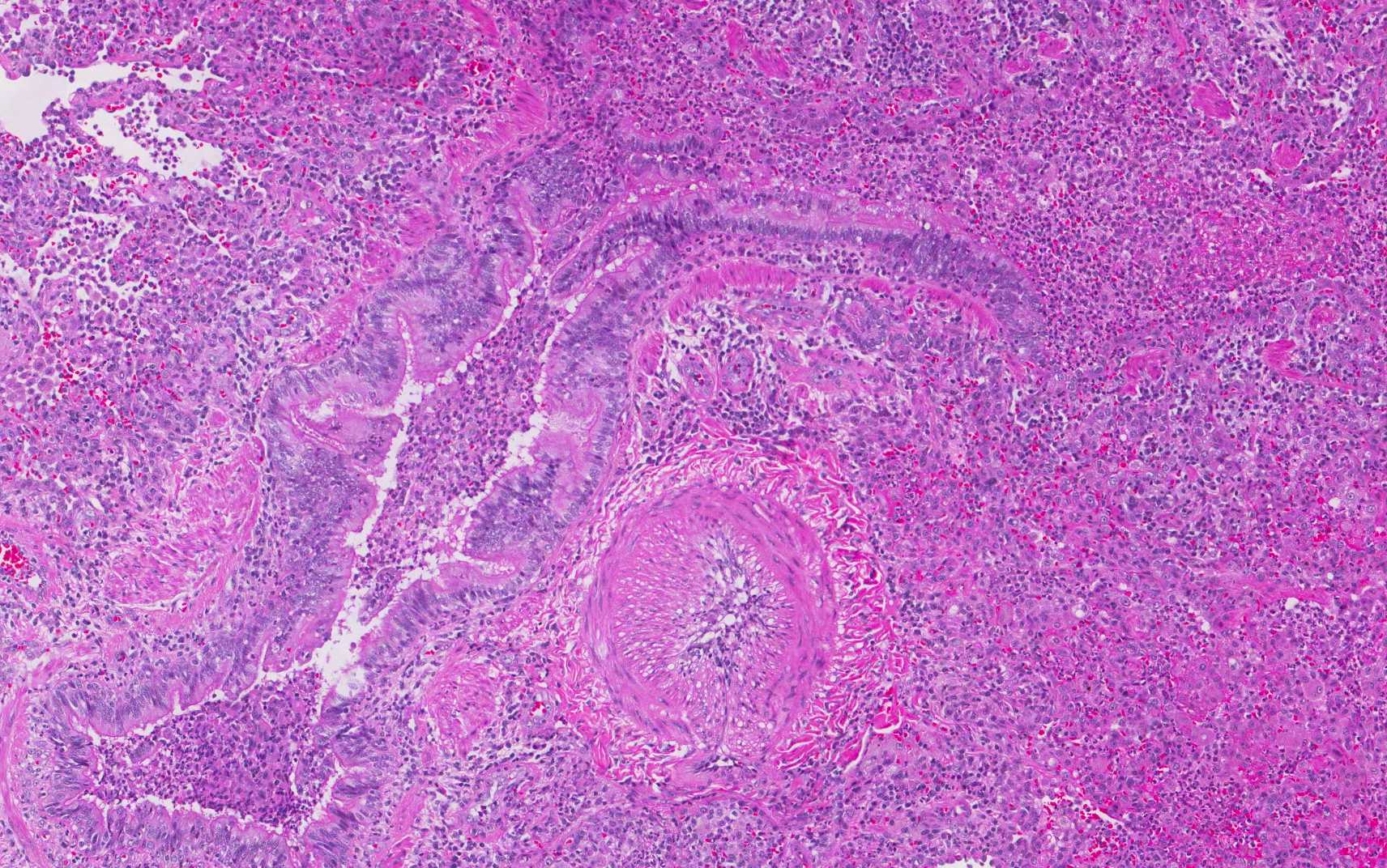
In addition, foci of bronchiectasis with parenchymal acute and chronic inflammation and interstitial fibrosis, classically reported in CF patients were noted (Figure 3). After lengthy discussion regarding the pathologic findings, her treating pulmonologist questioned the patient. She initially denied, but after extensive confrontation, eventually admitted to crushing and injecting alprazolam and hydrocodone/APAP via her central venous access.
DiscussionTop
Cystic fibrosis is a genetic autosomal recessive life-threatening disease that affects 30,000 people in the United States with 1000 new cases being diagnosed each year [7, 8]. It is caused by the presence of both copies of the gene for the CF transmembrane conductance regulator protein located at the q31.2 locus of chromosome 7 [9, 10]. Patients with CF have mucus build up in their lungs, pancreas, liver, intestines and other organs. In the lungs, mucus clogs the airways trapping bacterial, fungal and/or viral organisms leading to frequent infections, pneumonias, with subsequent lung inflammation, bronchiectasis and fibrosis [1, 2]. Other manifestations include sinus infections, failure to thrive, malnutrition, gastrointestinal symptoms and fatty stool secondary to pancreatic damage, diabetes, clubbing of fingers and toes, and infertility. While there is no cure for CF, the most important aspect of therapy is limiting and treating lung damage. This usually requires the repeated aggressive IV and oral antibiotics, daily mechanical airway clearance and inhalation of medicines to loosen and expectorate thick mucus, in addition to pancreatic enzyme supplements and multivitamins.
Cystic fibrosis is a chronic debilitating disease that significantly impacts the quality of life. Many of patients need more attention to their psychosocial needs in addition to their organic disease. The daily therapeutic routine, while effective, can be extremely time-consuming, frustrating and with added risks of complications. Several investigators have documented the use of recreational use of psychoactive drugs by patients with CF [11, 12].
The nonmedical use of prescription drugs has been cited as epidemic in the United States. The prevalence of prescription drug abuse has been on the rise in the last two decades, with 600,000 individuals reported first time users of pain relievers in 1990, up to 2.4 million in 2010 [13]. The highest rates are reported to be among adolescents and young adults [13]. A popular form of such abuse is the IV injection of solubilized, crushed or pulverized oral pain relief tablets. Rarely, it has been described in patients with history of parenteral nutrition or port-a-cath. Talc particles migrate through vessel walls to perivascular and pulmonary interstitial tissue; get phagocytosed by macrophages with giant cell reaction. Eventually these discrete small lung granulomatous nodules between 2-3 mm in diameter slowly replace the normal lung parenchyma resulting in extensive pulmonary fibrosis and pulmonary hypertension [14-16]. Classic histopathologic findings include the significant obliteration of the pulmonary vasculature by the embolizing foreign material with a florid giant cell reaction. Abundant needle-like or plaque-like talc crystals are easily identified within the granulomatous areas, spilling into the lung parenchyma inducing extensive damage and fibrosis. They are anisotropic when examined under polarized light [14-16].
A thorough clinical history and careful pathologic analysis are necessary to make the diagnosis of talc induced pulmonary granulomatosis. Because of the many overlapping clinical and radiological features between talc granulomatosis and pulmonary manifestations of CF, accurate diagnosis is challenging and requires a very high index of suspicion. Individuals with talc granulomatosis have variable clinical presentations depending on the severity of the disease. Patients usually present with nonspecific pulmonary symptoms including dyspnea, cough and increase in sputum production. However, a spectrum of changes could be seen varying from totally normal findings to the more severe conditions such as acute pulmonary hypertension due to pulmonary arterial occlusion, chronic pulmonary hypertension, emphysema, spontaneous pneumothorax, progressive massive fibrosis and acute respiratory distress syndrome. Imaging studies are helpful in identifying the widespread well-defined 2-3 mm nodules and a diffuse ground glass pattern. However they remain to be challenging due to the overlapping features with other granulomatous lung diseases, pulmonary hypertension, emphysema, infections and interstitial fibrosis in advanced conditions. CT pulmonary angiography is usually performed to rule out thromboembolic disease. Other than an open lung biopsy for confirmation of the diagnosis, many laboratory tests, pulmonary function tests, bronchoalveolar lavage, other imaging studies, echocardiography and heart catheterization have been proven to be of minimal value.
Cessation of IV drug abuse is of paramount importance for the appropriate management of these patients. Some investigators suggested the administration of prednisone in an attempt of reducing the pulmonary inflammatory reaction to improve blood flow and reduce pulmonary hypertension. Otherwise there is no clearly established treatment for the disease. Our patient was placed on prednisone 1mg/kg and tapered over 3 months. She had no radiographic response to steroid therapy. Her lung function eventually returned to her prior range and she was continued on her chronic CF therapies without central IV access. Eventually, she was treated for pulmonary hypertension with oral Adcirca with improvement in breathless and functional status.
Talc induced pulmonary granulomatosis in CF is indeed a rare finding. Only one documented case has reported thus far in the literature [16]. The accurate diagnosis of such condition has important clinical and social implications in patients with CF.ConclusionTop
Our goal was to increase the awareness of such under recognized potential cause of CF related pulmonary induced fibrosis and/or hypertension. Patients with risk factors and with findings similar to the reported patient in this report may benefit from further clinical and pathological evaluation for talc induced pulmonary granulomatosis.
Conflicts of interest
Authors declare no conflicts of interest.
ReferencesTop
[1]Flume PA. Pulmonary complications of cystic fibrosis. Respir Care. 2009; 54(5):618–627.Article Pubmed
[2]Ng MY, Flight W, Smith E. Pulmonary complications of cystic fibrosis. Clin Radiol. 2014; 69(3):e153–e162.Article Pubmed
[3]Chute DJ, Rawley J, Cox J, Bready RJ, Reiber K. Angiocentric systemic granulomatosis. Am J Forensic Med Pathol. 2010; 31(2):146–150.Article Pubmed
[4]Pintado V, Valencia ME, Lavilla P, Lopez-Dupla JM, Llanos C, et al. Angiothrombotic pulmonary granulomatosis in intravenous drug addicts. Rev Clin Esp 1991; 188(7):362–364.Pubmed
[5]Low SE, Nicol A. Talc induced pulmonary granulomatosis. J Clin Pathol. 2006; 59(2):223.Article Pubmed
[6]Farber HW, Falls R, Glauser FL. Transient pulmonary hypertension from the intravenous injection of crushed, suspended pentazocine tablets. Chest. 1981; 80(2):178–182.Article Pubmed
[7]O'Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009; 373(9678):1891–1904.Article Pubmed
[8]Ong T, Ramsey BW. Update in Cystic Fibrosis 2014. Am J Respir Crit Care Med. 2015; 192(6):669–675.Article Pubmed
[9]Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical applications. Nat Rev Genet. 2015; 16(1):45–56.Article Pubmed
[10]Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000; 67(2):117–133.Pubmed
[11]Stern RC, Byard PJ, Tomashefski JF Jr, Doershuk CF. Recreational use of psychoactive drugs by patients with cystic fibrosis. J Pediatr. 1987; 111(2):293–299.Pubmed
[12]Valencia LS, Cromer BA. Sexual activity and other high-risk behaviors in adolescents with chronic illness: a review. J Ped Adolesc Gynecol. 2000; 13(2):53–64.Article Pubmed
[13]Substance Abuse and Mental Health Services Administration, Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-48, HHS Publication No. (SMA) 14-4863. Rockville, MD: Substance Abuse and Mental Health Services Administration, 2014.Article
[14]Roberts WC. Pulmonary talc granulomas, pulmonary fibrosis, and pulmonary hypertension resulting from intravenous injection of talc-containing drugs intended for oral use. Proc (Bayl Univ Med Cent). 2002; 15(3):260–261.Pubmed
[15]Low SE, Nicol A. Talc induced pulmonary granulomatosis. J Clin Pathol. 59(2):223.Article Pubmed
[16]Smith KJ, Elidemir O, Dishop MK, Eldin KW, Tatevian N, et al. Intravenous injection of pharmaceutical tablets presenting as multiple pulmonary nodules and declining pulmonary function in an adolescent with cystic fibrosis. Pediatrics. 2006; 118(3):e924–e928.Article Pubmed
 Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved