


- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Modern Human Pathology
Volume 1, Issue 6, October 2016, Pages 44–62
ReviewOpen Access
Roles of HMGA proteins in cancer: Expression, pathways, and redundancies
-
Vincenzo Giancotti1,*
 ,
Palmina Cataldi2and
Claudio Rizzi2
,
Palmina Cataldi2and
Claudio Rizzi2
*Corresponding author: Vincenzo Giancotti, Department of Biochemistry, Biophysics and Macromolecular Chemistry, University of Trieste, Via L. Giorgeri 1, 34127 Trieste, Italy. Tel.: +39 0432920488; E-mail: giancotti@alice.it / giancot@units.it
Received 27 July 2016 Revised 15 September 2016 Accepted 21 September 2016 Published 28 September 2016
DOI: http://dx.doi.org/10.14312/2397-6845.2016-8
Copyright: © 2015 Giancotti V, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
The expression of the High Mobility Group A (HMGA) proteins, their participation in cancer signalling pathways, and their redundant functions have been reviewed in seven types of cancer: breast, colorectal, prostate, lung, ovarian, thyroid, and brain. The analysis of cell lines and tumours revealed an elevated level of their expression in all fully transformed cancer systems, which represents a step of the main cancer signalling pathways. In breast, colorectal, prostate, and lung cancers Wnt/β-catenin pathway is a master inducer of cell transformation in which are deeply involved HMG A1 and A2 proteins. On the other hand, IL-6/Stat3 pathway is responsible for cancer transformation in breast, lung, and prostate. The expression of HMGA1 in lung and ovarian cancers is due to an active PI3K/Akt pathway. The let-7 family of microRNA represses the expression of HMGA showing specificity by its different forms: the let-7b form is able to inhibit both proteins A1 and A2, the last also inhibited by a, c, d, and g forms. Moreover, both proteins are down-regulated by the repressor couple p53/microRNA-34a. The protein A1 and A2 participate to the Epithelial-Mesenchymal Transition cooperating with the three couples of factors Twist1/2, Snai1/2, and Zeb1/2. Through a combination of pathways, there is the simultaneous presence of high levels of both A1 and A2 together with the expression of other factors: a high co-operating efficiency is reached that supplies the tumour cells with properties of self-renewal, resistance, and invasiveness.
Keywords: HMGA; cancer signalling pathways; miR-let-7; Lin28; EMT; self-renewal
IntroductionTop
In the years between 1983 and 1987, four papers reported the identification of seven new low-molecular weight DNA-binding proteins that were highly present in the nucleus of cancer cells and absent in their normal counterparts. Altogether, they were named high mobility (HM) because of their high electrophoretic migration [1-4]. The following sequence determinations [5, 6] showed that the new protein group (G) actually only included three proteins: HMGI, HMGY, and HMGI-C. Later, to more rationally organise the nomenclature, including the other HMG proteins, the protein trio was re-named HMGA1a (HMGI), HMGA1b (HMGY), and HMGA2 (HMGI-C). Together, HMGA is one sub-class, while HMGB and HMGN are the other sub-classes [7]. We refer to HMGA1a and HMGA1b together as HMGA1 because they are derived from the same gene by alternative splicing. Altogether, the three proteins are called HMGA; however, we also referred to them as HMGA1/A2 in this report to make it clear that they are derived from two different genes. Since the first studies, it was clear that there was a link between their expression and both embryonic development and neoplastic transformation because, following virus infection, their onset was accompanied by a re-programming process of de-differentiation of the differentiated rat thyroid epithelial cells towards an undifferentiated phenotype together with the acquisition of tumour characteristics of growth [3, 4]. The aim of this review was to provide evidence of the properties of cells over-expressing HMGA and the cellular pathways in which they directly or indirectly participate. HMGA1 and HMGA2 have approximately 50% sequence homology, which includes three positive DNA-binding segments (called AT-hooks) and the C-terminal negative tail (Figure 1) [5, 6, 8-11]. Through protein/DNA and protein/protein interactions, they organise the stereospecific assembly of macromolecular complexes at the level of promoters/enhancers influencing gene transcription; however, they also affect the global organisation of the chromatin. Because of the shared properties mentioned above, they are considered fundamentally interchangeable.

In this review, we evaluate the latest findings, organising the illustration by groups of single cancer-types in which HMGA1 and HMGA2 are discussed together. Only a subset of all studied cancers (breast, colorectal, prostate, lung, ovarian, thyroid, and brain), for which we identified connections/similarities that are useful for common conclusions, is reported. However, when it is necessary to consider a broader perspective, a digression concerning some results found for other cancers is introduced. Among other data, it resulted that the Wnt/β-catenin pathway is always critically involved in the renewal, invasiveness, and epithelial-mesenchymal transition (EMT) of cancer cells. Moreover, an active Wnt/β-catenin pathway co-operates in cancer and stem cells with other pathways such as TGF-β and MEK-ERK pathways, and important factors such as Sox-2 and cMyc are present in both types of cells as well [12-15].
Breast cancer Top
The MDA-MB-231 cell line is a human basal-like triple negative (Er-, Pr-, Her2-) breast cancer (TNBC) cell line that is tumorigenic in immunodeficient mice and is characterised by high levels of both invasion and metastatic potential. This cell line was used in six studies we are going to discuss [16-21].
In the paper by Wend et al. [16] the elevated expression of HMGA2 in tumours and mouse embryos is related to activation of the Wnt10B/β-catenin pathway. In TNBC tumours (and cell lines), the Wnt pathway is active: β-catenin, free from its penta-degrading complex (β-catenin, CKIα, APC, Axin 1, and GSK-3β) moves into the nucleus where, together with Lef/Ctf factors, it induces the expression of HMGA2. There is the development of early stages of embryonic mammogenesis, cell proliferation (via cell cycle activation by cyclin A2), and the possibility of the prediction of relapse-free-survival and metastasis in TNBC patients. Another paper [17] also studied HMGA2 in MDA-MB-231 cells, among other systems. Here also, the expression of HMGA2 is found to be the result of activation of the Wnt/β-catenin pathway. HMGA2 expression, however, is tightly related to the expression of the Lin28 factor that is located downstream of Ctf/Lef activation, but upstream of HMGA2 production. In other words, this paper completes the observation of the preceding paper because it demonstrates that, following β-catenin/Ctf/Lef active onset, there is a sequence of events that includes the Lin28 protein, two members of the let-7 family of microRNAs (miRs) (let-7a and let-7f), and HMGA2. The binding of the Lin28 protein to let-7 induces the degradation of these miRs, which promote the expression of HMGA2 because let-7s are HMGA2 repressors, leading to cancer cell expansion. The paper of Guo et al. [18] confirms the Lin28/let-7/HMGA2 axis highlighted by Cai et al. [17], but its activation is the result of another pathway: oncostatin M (OSM)/JAK/Stat3. OSM is a pro-inflammatory cytokine belonging to the interleukin-6 (IL-6) family that induces the tyrosine phosphorylation of Stat3 (by JAK Janus Kinase and others) by binding to its receptor IL-6R in complex with the gp130 protein. This event results in the nuclear localisation of Stat3 that on the one hand induces the Lin28/let-7/HMGA2 axis (by blocking let-7), and, on the other hand, activates the expression of the factor ZEB1 by blocking another microRNA, i.e., miR-200. This work shows a twofold additional reason of interest: HMGA2 expression is related to the stromal microenvironment status (cells and cytokines) and explicitly links HMGA2 to the EMT together with ZEB1. It is worth noting that EMT is also promoted by the TGF-β/Smads and Ras/Raf/MAPK pathways [22-25].
The paper by Shah et al. [19] studied HMGA1 in MDA-MB-231 cells which, as reported by the authors, show stem-like properties due to the expression of HMGA1. Silencing of the HMGA1 gene involves the differential expression of 63 genes, most of which are expressed in embryonic tissues and are involved in cellular development. Moreover, HMGA1 silencing inhibits the stem-like properties of MDA-MB-231 cells and allows for reprogramming towards a more differentiated phenotype; consequently, silencing blocks mammosphere formation. HMGA1 enhances stem cell pluripotency by inducing the expression of cMyc, Sox-2, and Lin28; there is a loop by which cMyc in turn induces HMGA1 expression [19, 26]. Moreover, HMGA1 also induces the Stat3 gene in lymphoid tumorigenesis by binding to its promoter [27]. More recently, the clinical relevance of activated Stat3 signalling has been shown in diffuse large B-cell lymphoma [28]. Through Stat3, it is then possible to find a link between Stat3 and both HMGA1 [19] and HMGA2 expression [18] at least in TNBC cells; however, HMGA1 appears to be located upstream of Stat3, whereas HMGA2 is located downstream of Stat 3.
Other two papers also describe HMGA1 in breast cancer: the HMGA1 gene was silenced in MDA-MB-231 cells. The work by Di Cello et al. [20] reports that the knockdown of HMGA1 inhibits both anchorage-independent growth and tumoursphere formation. At the same time, tumorigenesis and metastasis are impaired in immunodeficient mice. The HMGA1 silencing induced in MDA-MB-231 cells by Pegoraro et al. [21] resulted in reduced malignant features of human breast cancer cells and inhibition of their migration and invasion both in vitro and in vivo. Similar to the work of Shah et al. [19], a HMGA1 gene expression signature, enriched in genes critical for migration, EMT and stemness, has been identified. The expression of HMGA1 activates aggressiveness-related and stemness-associated factors, and, conversely, HMGA1 silencing reverts the phenotype of TNBC cells according to the findings of Shah et al. [19]. Pegoraro’s paper highlights another link between HMGA1 and HMGA2 expression involving the Wnt/β-catenin pathway. Indeed, Wend et al. [16] and Cai et al. [17] suggest that this pathway is a signal that allows for the expression of HMGA2, whereas Pegoraro et al. [21] suggest that HMGA1 is an inducer of EMT/stemness via the key factors Lef1 and SETD8 of the Wnt/β-catenin pathway in the context of the HMGA1 130 gene signature. It is worth mentioning that the authors include the Notch pathway, which also promotes stemness, EMT, and metastasis in the same gene signature. The Notch pathway is linked to TGF-β/Smads, a well-known pathway that HMGA2 participates in [25], and also to the Wnt/β-catenin pathway, which is widely reported here. TGF-β is the major inducer of EMT, and cooperates with other pathways such as Wnt, Ras/Raf, Hedgehog, and Notch to induce EMT [22-24, 27-29].
The breast cancer papers discussed above allow us to conclude that both HMGA1 and HMGA2 are involved in the Wnt/β-catenin and IL-6/Stat3 pathways but in different ways: while HMGA2 is the result of active signals, HMGA1 appears to be a promoter of the signals. Consistent with this, the role of HMGA1 was previously studied by Treff et al. [30], who found that HMGA1 is related to another pathway, i.e., Ras/ERK. HMGA1 in breast cancer MCF-7 cells regulates genes involved in the extracellular activation of receptor tyrosine kinases (RTKs) [24] which activate the Ras/ERK cascade. Because the inhibition of Raf kinase (belonging to the same pathway) suppresses HMGA2 overexpression through Lin28 and let-7 [22], we speculate that HMGA1 could regulate HMGA2 through this pathway as well.
Figure 2 shows a scheme of the relationships that link HMGA1/A2 to pathways and cancer-related factors according to the discussed papers. The parallel Table 1 summarises the underlined properties of MDA-MB-231 cells and tumours highlighted by the cited authors and related to HMGA1/A2 expression/silencing. It is apparent that the overlap between HMGA1 and HMGA2 is large. Cell proliferation has been linked to the presence at least one of the proteins, as has aggressive tumour progression and metastasis. At the same time, HMGA1 and HMGA2 are important factors that induce EMT which is accompanied by stem-like properties including self-renewal and phenotypic reprogramming. The conclusion is that the over-expression of one of the HMGA1/A2 proteins in breast cancer cell lines or tumours is involved in the same effects. We cannot conclude that their presence at the chromatin level is redundant because they appear to be involved in different points in the transforming pathways and likely bind to different regions at the chromatin level. Although their concomitant presence may not be necessary, it could be relevant in tumour progression.

| References | HMGA1 | HMGA2 |
| Shah et al. [19] | Wend et al. [16] | |
| Di Cello et al. [20] | Cai et al. [17] | |
| Pegoraro et al. [21] | Guo et al. [18] | |
| Properties | 1. Tumour progression | 1. Tumour progression; Tumour expansion |
| 2. Stem-like and cancer stem cell properties | 2. Stem cell expansion | |
| 3. Self-renewal | 3. Self-renewal | |
| 4. Mammosphere formation | 4. Mammosphere formation | |
| 5. Epithelial-mesenchymal transition (EMT) | 5. Epithelial-mesenchylal transition (EMT) | |
| 6. Phenotypic reprogramming Phenotypic reversion | 6. Phenotypic reprogramming | |
| 7. Metastatic progression | 7. Metastatic prediction | |
| 8. Tumorigenic properties | 8. Cell migration and invasion | |
| 9. Cell migration and invasion | ||
| Pathways | A. IL-6/Stat3 pathway | A. IL-6/Stat3 pathway |
| B. Wnt/β-catenin pathway | B. Wnt/β-catenin pathway |
Colorectal cancerTop
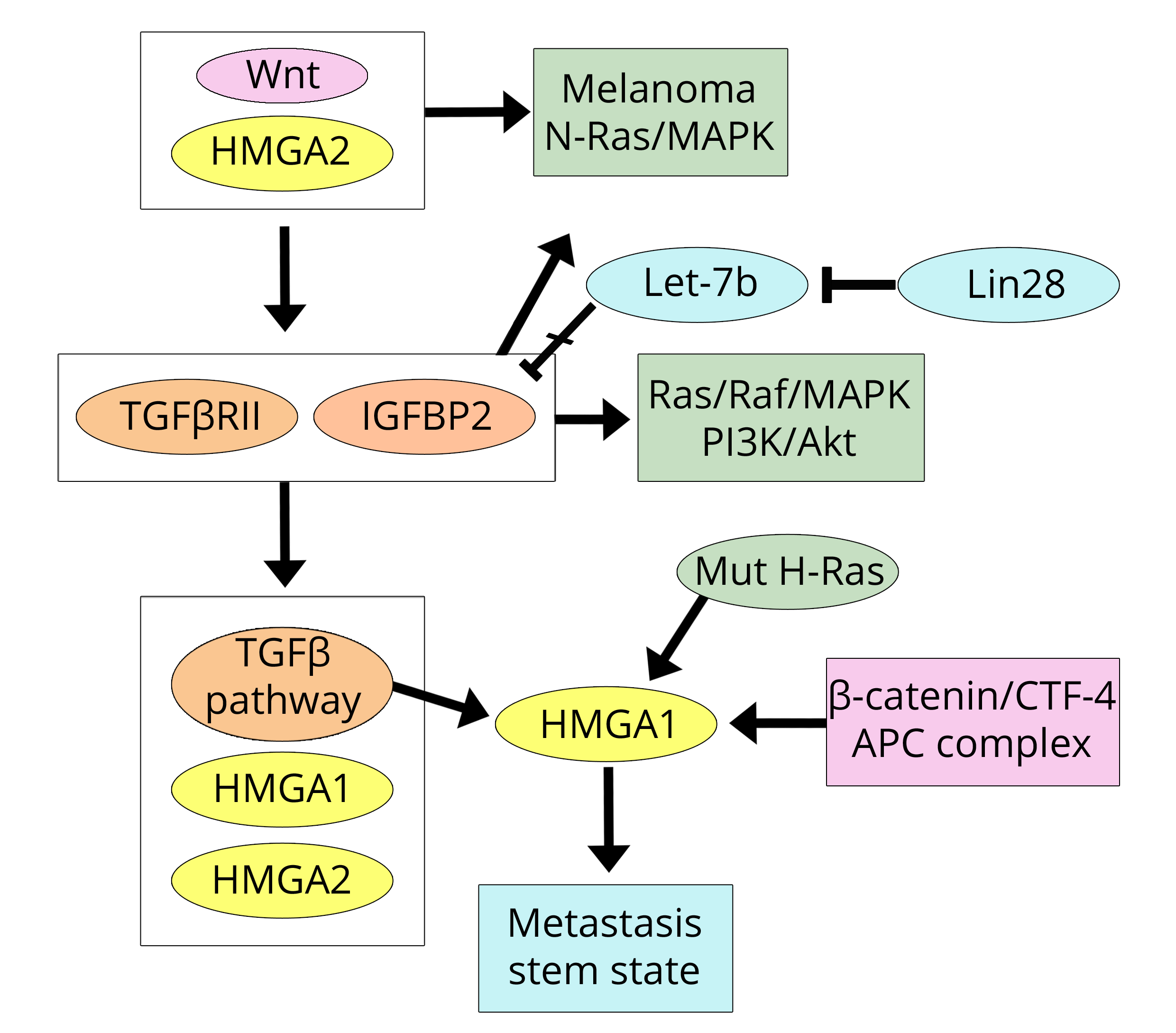
The MDA-MB-231 cells were used in the paper by Morishita et al. [31] due to their properties of high invasiveness related to the expression of the HMGA2 protein; this was a study concerning the acquisition of metastatic characteristics in both breast and colorectal cell lines and tumours. There is a clear consistency between this work and the previously discussed papers on breast cancer [16, 17] because Morishita et al. show that the Wnt pathway is active and initiates tumour onset in models of breast and colorectal cancer (CRC). Importantly, the presence of HMGA2, due to the active Wnt pathway, increases the expression of TGFβRII (TGF-β receptor II) and IGF2BP2 (Insulin-like growth factor 2, IGF2; m-RNA binding protein 2, BP2) therefore activating the TGF-β pathway (former factor) and both the Ras/Raf/MAP and PI3K/Akt (latter factor) pathways. In other words, the Wnt/HMGA pathway establishes a triplicate connection with the three pathways induced by the factors IGF, EGF, and TGF-β [24]. This allows for the inevitable progression and invasiveness of the tumour initiated via Wnt. It is worth noting that, in the paper under discussion, elevated expression (mRNA) of HMGA1 was also reported in five human cancer cell lines; however, all of the work was focused on HMGA2. Regarding this protein, the authors declare that “HMGA2 is upstream of the TGF-β pathway” probably because HMGA2 has previously been induced by the Wnt pathway. However, in other studies, it has also been reported [25] that the TGF-β pathway induces HMGA2 expression, which then localises downstream of the TGF-β pathway. It seems likely that in addition to the existing HMGA2 derived from the activated Wnt pathway, another output is added once TGF-β is activated. Although the relationship between HMGA2 expression and that of IGF2BP2 reported by Morishita et al. [31] is very clear, the mechanism of regulation could be more complex because Alajez et al. [32] reported that in head and neck cancer, the increase in IGF2BP2 is due to Lin28B, which represses let-7b, and allows its expression. Moreover, the HMGA2 location is further questionable, as indicated by the results reported by Zha et al. [33] in gastric cancer. Indeed, in this study, it is shown that HMGA2 activates the Wnt/β-catenin pathway rather than representing a product of this pathway. In conclusion, the paper by Morishita et al. [31] highlights three important points: (i) the Wnt pathway activated in breast cancer, and activating the expression of the HMGA2 protein, is also common to CRC; (ii) there is crosstalk between the Wnt and TGF-β pathways; and (iii) the concert between these two pathways allows tumour progression and stroma invasion characterised by the presence of HMGA2 (and TGFβRII and IGF2BP2) in the tumour cells of the invasive front, as also demonstrated in another study on CRC [34]. Stromal invasion by tumour cells means that these cells have undergone the EMT in which the TGF-β pathway and related HMGA2 expression have been well documented [25]. The HMGA2-IGF2BP2 relationship deserves further attention. IGF2BP2 was originally reported as a factor targeted by HMGA2 in mouse embryonic development [35] and subsequent studies have demonstrated that HMGA2/IGF2BP2 also regulates oncogenesis [36], particularly by activation of the NRAS/MAPK pathway. The relationship with NRAS directs us towards a field of great interest because NRAS is related to melanoma and recent data [37] link HMGA2 to NRAS and melanoma. Moreover, a link between the HRAS and HMGA proteins in colon cancer was previously stated by Cleynen et al. [38]. In the human colon cancer cell line HCT116, HMGA1 is induced by mutant H-Ras oncogene via promoter regions regulated by SP1 (Specificity Protein 1) and AP1 (Activator Protein 1). Additionally, the HMGA1 protein is barely observable in WT-Ras cell lines (such as Colo-205 and Caco-2), but is detectable at high levels in mutant H-Ras cell lines (such as DLD-1, HCT116, SW480, and HT29).
While the paper by Morishita et al. [31] confirms the Wnt/HMGA2 axis in breast cancer according to Wend et al. [16] and Cai et al. [17], it emphasises its action in colorectal cancer. This conclusion can be linked to the other protein (HMGA1), as found in colon cancer by Bush et al. [39]. Indeed, Bush et al. demonstrated that the TCF-4 factor, belonging to the Wnt/β-catenin pathway, binds to the human HMGA1 promoter and up-regulates HMGA1. The colon cancer picture is completed by the cited findings of Cleynen et al. [38] and by the work of Belton et al. [40] that demonstrated that HMGA1 regulates metastatic progression in colon cancer by maintaining a stem-like state of the cells. In Figure 3, the actions of the trio HMGA2/TGF βRII/IGFBP2 in CRC are shown, along with the link with breast cancer.
Prostate cancerTop
In prostate cancer, Wnt is also suggested to be the pathway responsible for the initiation and progression of cancer, as found for CRC, and this event is mediated by HMGA2. In the paper by Zong et al. [41], Wnt induction is shown to be a result of the presence of HMGA2 in the microenvironment of mesenchymal stromal cells that, because of the effect of HMGA2, secrete factors with a paracrine action on prostatic epithelial cells, thereby inducing cancer initiation. Together with the IL-6 effect [18], there is also another result that highlights an interaction/cooperation linking molecular events of the microenvironment to HMGA proteins and cell transformation. Another aspect of the HMGA/prostate relationship involves the HMGA1/let-7 axis shown in the Schubert et al. paper [42]. In this paper, it is shown that reduced expression of let-7b induces an increase in the expression of HMGA1 because let-7b targets the 3’UTR of HMGA1. Down-regulation of let-7b is associated with aggressive cancers and can be considered an independent prognostic marker. Actually, the paper by Schubert et al. [42] includes not only let-7b miR, but also two other members of the let-7 family (a and c), together with other miRs that the authors demonstrated to be down-regulated in prostate cancer. Because this review addresses the regulation and function of HMGA1/A2, we are only going to consider let-7a/b/c, among which the b-form (according to the paper under discussion) is strongly related to prostate cancer and HMGA1 down-regulation. However, in a previous paper by Wei et al. [43], it was reported that HMGA1 is also down-regulated in the prostate by miR-296 but not by let-7c. Conversely, let-7c down-regulated HMGA2 which is not down-regulated by miR-296 (Figure 4). The differential regulation of HMGA1/A2 by miRs could be due, at least partially, to these findings.

Active androgen receptor (AR), in its active state due to the binding of androgens (A) (such as dihydrotestosterone), is translocated from the cytoplasm into the nucleus where its target genes are regulated. Androgen deprivation is the choice therapy, but its effectiveness is time-limited because an androgen-independent mechanism of DNA transactivation arises that is characteristic of metastatic castrate-resistant prostate cancer (mCRPC) [44]. Among others, two events could rescue the androgen deprivation: increased miR-21 expression, associated with a malignant state, and loss of PTEN, associated with increasing activity of the PI3K/Akt pathway [45]. Therefore, it is not surprising that Takeuchi et al. [46] report that HMGA1 is induced by androgen-deprivation in prostate tumour cells. In addition to the PI3K/Akt pathway, there are other pathway changes by which HMGA1 could be expressed in prostate cancer: through a Ras initiating pathway [38] or by a pathway including let-7, such as Wnt or IL-6/Stat3, in which repressed let-7 causes HMGA1 expression [38, 42, 43] (Figure 4). The IL-6/JAK/Stat3 pathway is active in both CRC and prostate cancer, suggesting that its pathway factors could be considered therapeutic targets [47]. Moreover, Stat3 signalling in prostate cancer is able to integrate different signalling pathways and, in androgen deprived cases, to re-activate AR nuclear activity [48]. TGF-β1 stimulates Stat3 phosphorylation that, in turn, by binding to the Twist1 promoter, induces its expression and consequently prostate cancer invasion, as Twist1 is one of the main regulators of EMT [49]. However, TGF-β1 phosphorylated Stat3 (pStat3), as shown in Figure 2, belongs to the IL-6 pathway of breast cancer from which HMGA2 is derived. As was very recently reported [50], TGF-β1 induces HMGA1 via Sp1 right in MDA-MB-231 breast cancer cells. TGF-β1 then directly induces both HMGA1/A2 [25, 50], and indirectly induces HMGA2 by pStat3 of the IL-6 initiating pathway.
All of these data on the Stat-3 pathway converge in the paper by Rokavec et al., [51] in which a triplicate study was carried out that includes the three cancers that have been discussed so far: breast, CRC, and prostate. This study revealed that, in the three cancers, EMT, invasion, and metastasis are the result of IL-6R/Stat3 pathway activation and, importantly, added another piece of information of great interest to the regulation of this pathway. Indeed, a feed-back loop due to miR-34a that suppresses the IL-6R/Stat3 pathway by down-regulating IL-6R expression has been demonstrated, and active p53 is responsible for inducing miR-34a. The function of p53 in regulating cancer involving miRs (including the miR-34 family) has already been discussed in another review [52] and since 2008, it has been shown that all forms of miR-34 (a, b, and c) repress the expression of HMGA2 in gastric cancer [53].
Another indirect, but very significant link, between Stat3 and HMGA2 can be found by overlapping the results of the paper by Yousuf et al. [54] with those by Yun et al. [55] while also considering the papers by Sun et al. [56] and Dangi-Garimella et al. [22] at the same time. RKIP, the Raf kinase inhibitor protein, is able to inhibit the multi-phosphorylation of Stat3 not only by Raf, but also by c-Src and JAK in both breast (MDA-MB-231) and prostate cancer cells. As a result, nuclear translocation of Stat3 is prevented and, consequently, the effects of Stat3 on gene expression are abolished. Because of its anti-kinase activity, RKIP is considered a tumour metastasis suppressor that acts by inhibiting Lin28; this event is followed by the over-expression of let-7 (as we already reported), which in turn inhibits the expression of metastasis-promoting factors such as HMGA2 [22, 55]. Figure 5 shows p53, miR-34a, Stat3, and HMGA2 connections together with the action of RKIP on both Stat3 phosphorylation and Lin28 repression.

Lung cancerTop
The expression of HMGA1/A2 proteins was thoroughly investigated in non-small-cell lung cancer (NSCLC), which includes most lung tumours (adenocarcinoma, squamous-cell carcinoma, and large-cell lung cancer), by four groups from 2007-2009 [57-60]. In three of these papers, HMGA2 was studied, whereas Hillion et al. [58] investigated HMGA1. The four studies were carried out in both humans and mice using tumour cell lines and primary tumours along with RT-PCR and immunohistochemistry to obtain data regarding protein expression. As a general rule, the proteins were highly expressed in all transformed cells and tumour tissues. Some details could be of interest. Regarding HMGA1, Hillion et al. [58] found that HMGA1 resulted in a transformed phenotype starting from normal tissue and, by binding to the promoter of MMP-2, positively affected its expression; together, these factors provide large-cell carcinoma cells with anchorage-independent cell growth (Figure 6a). The immunohistochemistry and RT-PCR studies by Meyer et al. [60], carried out in adenocarcinoma and squamous-cell carcinoma, related HMGA2 expression to tumour grade. Using a similar approach (similar techniques and cancer types), Di Cello et al. [57] confirmed that HMGA2 is correlated with tumour grade and also demonstrated that HMGA2 inhibition blocks the transformation. On the other hand, Kumar et al. [59] studied HMGA2 down-regulation by the let-7 family and found that let-7g expression induced both cell cycle arrest and cell death in adenocarcinoma mouse cells expressing mutant G12D-K-Ras (Figure 6a, b). The involvement of Ras-pathways with HMGA1/A2 proteins has already been reported in breast and CRC sections [30, 36-38]. Among the papers on NSCLC discussed above, the paper by Hillion et al. [58] is the only one that provides information about the origin of the HMGA production in lung cancer, in other words, about the pathway(s) that results in HMGA1/A2 expression linked to cell transformation. Involvement of the let-7 family in lung cancer, as reported by Kumar et al. [59], was subsequently confirmed by Wang et al. [61] although they related the decreased protein levels of both K-Ras and HMGA2 to the over-expression of the let-7a form (instead of the g form), which also inhibits the proliferation and the invasion of the NSCLC cell lines used. Conversely, it was reported more recently in hepatocellular carcinoma (HCC) cell lines and tumours that only let-7g inhibits many cancer properties [62] (Figure 6b). Indeed, among the let-7 family, let-7g has the lowest expression in HCC and its re-expression results in a variety of effects: inhibition of proliferation, migration, and invasion; induction of apoptosis; variation of the cell cycle; and suppression of EMT by the down-regulation of K-Ras/ERK1/2/HMGA2/Snail activity. Therefore, there is no doubt that there is a strong association between the let-7 family and HMGA2 in lung cancer, and, as already discussed in the Breast section, it is likely that, among all of the pathways in which the let-7 family participates, Wnt/β-catenin and IL-6/JAK/Stat3 are starting points in lung cancer as well (Table 1). Additionally, Stat3 could be the most involved factor in lung cancer, as a recent review [63] placed its phosphorylation at the intersection of many co-operating pathways such as IL-6/JAK, RTKs, Ras/Raf, PI3K/Akt, Src, and Abl. However, there are other miRs that can regulate the oncogenic properties of HMGA2 in addition to the let-7 family. Rice et al. [64], in a study on lung cancer cell lines, reported that the thyroid transcription factor-1 (TTF-1, also named NKX2-1) acts as an anti-metastatic factor because it activates miR-33a, which then represses the expression of HMGA2, as does let-7, but by binding to a different site of the 3’UTR of HMGA2. Regarding the PI3K/Akt pathway, it is worth mentioning the study of Scrima et al. [65], which demonstrated that the activation of PI3K contributes to cell proliferation and tumorigenicity of NSCLC cells; specifically, the activated PI3KCA catalytic subunit up-regulates oncogenic transcription factors such as HMGA1, Fos, and cMyc (Figure 6a, b).

Additionally, a consistent link between the two different pathways has recently been demonstrated: Stat3 blocking in NSCLC enhances the effect of the dual inhibitor BEZ235 on both PI3K and mTOR [66]. Indeed, many manufacturing companies have developed specific inhibitors that target members of the cited pathways in lung cancer such as RTK/EGFR (Erlotinib*, Roche), PI3K (BKM 120, Novartis), mTOR (Sirolimus/Rapamune, Pfizer), Akt (MK 2206, Merk), Raf (RAF 265, Novartis), and MEK (RO 4987655, Hoffman-La-Roche) [67].
Ovarian cancerTop
Although only a limited number of papers regarding the HMGA proteins can be found in the literature, we decided to discuss ovarian cancer as this cancer is of high clinical importance because its high-grade type is one of the most lethal among carcinomas. Ovarian cancers have an elevated variable morphology meaning that many different sub-types have been reported according to the origin of cells from the reproductive apparatus that initiate (presumably) the tumour transformation. The aim of this work is not to review the various and combined approaches (morphologic, genetic, and molecular) used to classify ovarian cancer, but rather to ascertain HMGA expression and its correlation with tumour onset and progression. Therefore, we are going to discuss a number of cases assuming tumour typing as the author suggested without any further comment. In many cases, we will refer to the dualistic model that classifies the various histological ovarian cancers into only two types, i.e., type I (low-grade) and type II (high-grade), which was introduced by Kurman [68] and Koshiyama et al. [69] because this is more useful for the molecular presentation we adopt and the two types are also very different from a genetic standpoint.
The paper by Mahajan et al. [70] is of twofold interest: (i) six different ovarian tumours were analysed (Clear Cell Ovarian Carcinoma, Endometrioid Ovarian Carcinoma, High-Grade Papillary Serous Carcinoma, Malignant Mixed Mullerian Tumour, Mucinous Ovarian Carcinoma, and Serous Borderline Tumour), although the number of cases was rather low (115 total); (ii) both HMGA1 and HMGA2 proteins were analysed by immunohistochemistry. It is possible to deduce from this work that: (i) all tumour samples express both HMGA1 and HMGA2 and, as shown within each HMGA1 or HMGA2 column of data in Table 2 (of the paper under discussion), different levels of HMGA1 and HMGA2 can be observed in positive cases, for each type of tumour, by number of cases and by the intensity of tumour immunostaining; (ii) HMGA1 is more frequently expressed than HMGA2 (85.2% versus 29.6% of cases, respectively), but the intensity of nuclear immunostaining revealed much higher fluctuation for HMGA2 than HMGA1. Indeed, considering the two most different intensities in the HMGA1/A2 columns (Table 2), it is possible to obtain a ratio maximum/minimum of 1.6 (Malignant Mixed Mullerian/Endometrial Ovarian carcinoma) for HMGA1 versus a ratio of 12.1 (High-grade Papillary Serous Ovarian Carcinoma/Mucinous Ovarian Carcinoma for HMGA2).
| (A) HMGA1 positivity/Total cases | |||||
| References | Chiappetta et al. [83] | Czyż et al. [84] | |||
| Specimen/technique | FFPE/IHC | Frozen/RT-PCR | |||
| Tissue | |||||
| (a) Normal thyroids, Goitres, Thyroiditis, Hyperplastic nodules | 1/32 | 0/17 | |||
| (b) Follicular Thyroid Adenoma (FTA) | 44/200 | 0/12 | |||
| (c) Follicular Thyroid Carcinoma (FTC) | 18/19 | 31/37 | |||
| (d) Papillary Thyroid Carcinoma (PTC), Follicular Variant Papillary Thyroid Carcinoma (FVPTC) | 92/96 | 0/11 | |||
| (e) Anaplastic Thyroid Carcinoma (ATC) | 11/11 | ----- | |||
| (B) HMGA2 positivity/Total cases. | |||||
| References | Belge et al. [91] | Chiappetta et al. [92] | Jin et al. [93] Lappinga et al. [94] |
Prasad et al. [95] | |
| Specimen | FFPE | FFPE/FROZEN | FFPE//FNA | FFPE | |
| Technique | qRT-PCR | IHC/qRT-PCR | qRT-PCR | IHC | |
| Tissue | |||||
| (a) Normal thyroids, Goitres, Thyroiditis, Hyperplastic nodules | 0/5 | 0/19 ----- | 0/6 0/28 | 1/36 | |
| (b) Follicular Thyroid Adenoma (FTA) | 5/19 | 3/31 1/7 | 1/34 4/55 | 0/30 | |
| (c) Follicular Thyroid Carcinoma (FTC) | 8/9 | 4/21 13/16 | 19/23 27/30 | 6/17 | |
| (d) Papillary Thyroid Carcinoma (PTC), Follicular Variant Papillary Thyroid Carcinoma (FVPTC) | 27/28 | 30/45 34/37 | 58/78 39/56 | 38/56 | |
| (e) Anaplastic Thyroid, Carcinoma (ATC) | 3/3 | 11/12 4/4 | 2/3 2/2 | ---- | |
Abbreviations: IHC: immunohistochemistry; qRT-PCR: quantitative real time polymerase chain reaction; FFPE: formalin-fixed paraffin-embedded; FNA: fine needle aspiration.
It appears that for the different types of ovarian cancer, both the expression and staining intensity of HMGA1 are approximately constant, whereas the HMGA2 trend is variable. The authors concentrated their attention on HMGA2, which is associated with an elevated number of positive cases and high nuclear staining in high-grade serous ovarian carcinoma, and they concluded that HMGA2 over-expression is a characteristic of type II ovarian tumours in the early stages. It should be mentioned that Mahajan and colleagues also examined the expression of some forms of the let-7 family. The results from let-7b are interesting: in high-grade papillary serous ovarian carcinoma (HG-PSOC), let-7b was significantly down-regulated, whereas, in endometrial ovarian carcinoma (EOC), no difference was noted. If we compare this result with Table 2 of the paper under discussion, we observe that the data of these two tumours indicate that HMGA1 expression (the percentage of positive cases) and nuclear intensity staining are essentially the same (85.7% versus 82.2, and 1.06 versus 0.91), respectively. The comparison of the HMGA2 data shows instead that both HMGA2 expression and staining intensity in HG-PSOC is much higher than in EOC (60.0% versus 6.6, and 1.57 versus 0.14, respectively), which is consistent with let-7b down-regulation in HG-PSOC. This means that let-7b selectively regulates HMGA2 in the different types of ovarian cancer rather than HMGA1. Some years before, Masciullo et al. [71] reported that HMGA1 was intermediately or strongly expressed in at least 80% of primary ovarian carcinomas. This result was confirmed by Peters et al. [72] in a study in which they compared gene expression in normal ovarian surface epithelium with various histological types of ovarian carcinomas. HMGA1 resulted in a cluster of over-expressed genes that have been suggested to be biomarkers of ovarian cancer. However, most of the studies on ovarian cancer investigated the HMGA2 protein. Since 2008, Malek et al. [73] demonstrated that silencing HMGA2, which is over-expressed in Ras-transformed rat ovarian surface epithelial cells, results in the inhibition of tumour growth and increased apoptosis (Figure 7a).
Consistently, inducing HMGA2 over-expression is sufficient for early transformation of ovarian surface epithelial cells, according to a process that targets genes involved in EMT [74]. Moreover, HMGA2 over-expression promotes both cell migration and formation of xenograft tumours. The expression of HMGA2 is also considered to be an early event in ovarian carcinoma by other authors who show that the expression is a common characteristic of different types of tumours, of the progression of the disease, and of EMT properties [75-77]. Consistently, Montserrat et al. [77], in endometrioid endometrial carcinoma, found that the whole set of E-cadherin repressors, i.e. SNAI1, TWIST, ZEB1, HMGA2, and SNAI2 (SLUG), is over-expressed in the most advanced tumours, and the over-expression of ZEB1, HMGA2, and SLUG is further increased at the myoinvasive front similarly to the invasive front of CRC [31, 34]. Moreover, the papers by Park et al. [76] and Montserrat et al. [77] also provide mechanistic data regarding the repression/activation of HMGA2 as an early transforming factor in ovarian cancers. On the one hand, the let-7 family of miRs targets and represses HMGA2 mRNA (as we reported above for other cancers and in this section for let-7b [70], while, on the other hand, B-Raf/MAPK/MEK/ERK is suggested to be the pathway that represses E-cadherin and activates the above cited EMT factors [77]; the mutation V600E of B-Raf is the constitutive activator of the process. As recently reported [78], the level of HMGA2 in serous ovarian carcinoma can be high not only if miR-let-7 family members are down-regulated but also if there is a shortening of the 3’UTR of the HMGA2 mRNA even if no decrease in let-7 is observed because there is a loss of binding sites for miRs. However, there are other miRs that regulate HMGA2 expression in high-grade serous ovarian carcinoma besides the well-known let-7 family. For example, miR-182 has an opposing action because its over-expression in advanced ovarian cancer is significantly associated with increased HMGA2 expression, enhanced tumour invasiveness, and metastasis [79, 80] (Figure 7b).

Among the ovarian cancer papers, there are two important studies regarding the involved transforming pathways that are related and mutually supportive of each other [81, 82]. One addresses HMGA1, while the other addresses HMGA2; the former indicates PI3K/Akt as the transforming pathway in ovarian cancer, while the latter shows the involvement of the KRas/Raf/MEK/ERK pathway, with Akt and Raf being the hub factors of the pathways, respectively. De Marco et al. [81] studied samples of human ovarian carcinoma and used cell lines as controls: most of the cases (79%) showed PI3K/Akt activation due to over-expression of the mutated PI3KCA subunit. PI3K/Akt signalling occurs early in the tumorigenic process that gives rise to the over-expression of the oncogenic factors HMGA1, Fos, Jun-B, and cMyc, as confirmed by experiments in cell lines. In the paper by Stelniec-Koltz et al. [82], rat ovarian surface epithelial cells were transformed by mutant K-Ras, which resulted in enhanced activity of the Ras/Raf/MEK/ERK pathway. The transformation of cells by mutant K-Ras was associated with the de-regulation of 1826 genes, of which 1562 were controlled by a network formed by only 7 over-expressed factors: Fosl1, HMGA2, Otx1, Klf6, Gfi1, Jun-B, and RelA. Therefore, both papers, using two different mutant agents, identify, in different pathways, factors that are subsequently able to manage the other events of cancer progression. At least one of these, i.e., Jun-B, is common to both pathways. However, another more direct link between the two pathways can be found in the work by Stelniec-Klotz et al. [82] because it demonstrated that, inhibiting PI3K in the KRas-transformed cells, the over-expression of HMGA2 is strongly reduced. This means that the two pathways, at least regarding HMGA2, are linked (Figure 7b).
Thyroid cancerTop
Because thyroid cells were the first system in which the HMGA proteins were identified and related to the onset of neoplastic transformation by retrovirus infection of differentiated cells [3, 4], it is understandable that the same research group carried out studies on thyroid tumours. Indeed, the study by Chiappetta et al. [83] addresses the HMGA1 (in this paper still named HMGI) expression in 326 thyroid tumours and 32 normal thyroid tissues and goitres. All of these samples were analysed by IHC, and some selected cases (31 tumours and 9 normal tissues and goitres) were also analysed by RT-PCR. Normal tissues and goitres exhibited fundamentally negative results: Only one positive goitre was identified by IHC, and no positivity was found by RT-PCR. Very high levels of IHC positivity were detected in all carcinomas: 95% follicular, 96% papillary, and 100% undifferentiated anaplastic tumours. These results were consistent with the RT-PCR analyses. Follicular adenomas showed an intermediate positivity averaging 22% by IHC and 40% by RT-PCR. It is worth mentioning that Chiappetta et al. [84] also analysed some fine needle aspiration biopsies (FNA): Ten cytologically diagnosed follicular neoplasms suspicious for follicular carcinoma and two papillary carcinomas. FNA analyses by both IHC and RT-PCR showed that 8 out of the 10 neoplasms were follicular adenomas (HMGA1 negative) and the remaining 2 were indeed follicular carcinomas detected as positive by both analyses. The two suspicious papillary carcinomas were found to be HMGA1 positive by both IHC and RT-PCR. However, the number of analysed samples is too low to reach a conclusive statement about the reported HMGA1/FNA data without any doubt.
Years later, another research group used RT-PCR to analyse 60 thyroid tumours and 17 normal thyroid tissues as controls [84]. Regarding the normal control tissues and follicular cancers, the results are rather similar to those of Chiappetta et al. [83] because normal tissues were HMGA1 negative, whereas the percentage of positive follicular carcinomas was very high (84%). All follicular adenomas were negative which is different from the previous result, which showed 22-40% of positivity. However, the result by Czyż et al. [84] regarding the 11 papillary cancers was strongly discordant because all were negative, whereas Chiappetta et al. [83] reported 96% positivity by IHC and 100% positivity by RT-PCR. Czyż and colleagues [84] also checked for the presence of HMGA1 mRNA in the blood of 17 patients diagnosed with papillary carcinoma and 6 patients diagnosed with follicular carcinoma. Only 1 out of the 17 papillary carcinomas resulted in HMGA1-positive blood, while 5 follicular carcinomas resulted in HMGA1-positive blood. Although this type of study is of unquestionable interest because of the importance of finding new markers in the blood, again the number of analysed cases is too low. We compared in Table 2 the HMGA1 expression data in thyroid cancer obtained by the two cited laboratories. The HMGA1 positivity of papillary thyroid carcinomas reported by Chiappetta and colleagues [83] was indirectly confirmed by the same research group in a subsequent study in which it is reported that 11 papillary thyroid carcinomas show HMGA1 positivity, although with variable degrees of intensity [85].
An inhibitory function of HMGA1 is reported by Frasca et al. [86] who showed a direct interaction with p53 that results in the inhibition of its suppressor activity in thyroid cancer cells. It was also demonstrated that HMGA1 is essential to reaching complete tumour transformation because virus-infected (v-Ras-Ki) thyroid cells in which the expression of HMGA1 has been blocked do not show the full malignant phenotype, although they are able to grow without hormone stimulation [87]. Indeed, it has been subsequently reported (in a different cancer) by Cleynen et al. [38] that HMGA1 should be present in an H-Ras transforming pathway (CRC section, Figure 3).
Another regulatory property of HMGA1 was demonstrated by the paper of Mussnich et al. [88] which studied its action on a group of miRs involved in thyroid cancer: miR-10b, miR-21, miR-125b, miR-221, and miR-222 (positively regulated by HMGA1) and miR-34a and miR-603 (negatively regulated by HMGA1). The authors focused their attention on miR-10b and miR-603, which both work toward the same end but the former is positively regulated by HMGA1 and the latter is negatively regulated by HMGA1. Indeed, miR-10b targets and represses PTEN, a tumour suppressor of the PI3K/Akt pathway, which we reported above to be one of the primary promoters of cancer onset and progression. Conversely, miR-603, because of the action of HMGA1, has a reduced effect on the CCD1 and CCD2 cyclins. Taken together, the result is cell cycle and tumour progression. In this paper, there is another point of interest. Among the other HMGA1-regulated miRs (all of elevated importance in cancer), miR-34a is of special interest because, as we reported in the prostate section, miR-34a is related to p53 and this has been studied by Frasca and colleagues [86] in thyroid cancer. Both p53 and miR-34a are onco-suppressors with the latter being under the action of the former. However, both are HMGA1-dependent: HMGA1 inhibits p53 [87] and negatively regulates miR-34a [89]. It is a triangle of interconnected factors of which one (HMGA1) is tumour promoting, while the others (p53 and miR-34) are tumour suppressors (Figure 8a). A similar relationship linking p53 and miR-34a can also be found for HMGA2, but this includes evidence from two other cancers, i.e., gastric and breast [89, 90] and includes Bcl-2 in the reciprocal connections. In gastric MKN-45 cancer cells, HMGA2 silencing induces apoptosis by repressing Bcl-2 (a known anti-apoptotic factor), while in breast MDA-MB-231 cancer cells the active miR-34a down-regulates Bcl-2 and increases apoptosis. Therefore, HMGA2 is anti-apoptotic (as is HMGA1), miR-34a is pro-apoptotic, and silencing the former has the same effect as an active miR-34a state. At the same time, while wild-type p53 activates miR-34a, in a mutant p53 system, the repressive effects of miR-34a on Bcl-2 are abolished (Figure 8b).

The expression in thyroid cancer of HMGA2 was analysed as well. Bedge et al. [91] analysed HMGA2 expression by qRT-PCR in 59 thyroid tumour samples: 19 adenomas, 9 follicular carcinomas, 28 papillary carcinomas, and 3 anaplastic carcinomas (Table 2; positivity numbers were obtained from Table 1 and Figure 2 of the authors’ paper). Five normal or thyroiditis samples were used as negative reference. At the same time, Chiappetta et al. [92] analysed 109 samples of FFPE (formalin-fixed paraffin-embedded) thyroid tumours by IHC: 31 adenomas, 21 follicular carcinomas, 45 papillary carcinomas, and 12 anaplastic carcinomas. A total of 19 samples from normal tissues or goitres were used as negative references. The same authors also analysed 64 frozen specimens by qRT-PCR (Table 2). Comparing the results of the two studies it is possible to conclude that the two types of analyses (RT-PCR and IHC) produced very similar results in that normal tissues, goitres, and thyroiditis were HMGA2 negative, whereas, anaplastic carcinomas were 100% positive. However, variable expression, at high levels of positivity, was observed in papillary carcinoma, from 67% by IHC in Chiappetta et al. [92] up to 96% by qRT-PCR in Bedge et al. [91]. Chiappetta’s IHC data showed the lowest percentage; on the contrary, the qRT-PCR percentages were very similar (96% versus 92%). As expected, HMGA2 expression in adenomas was rather low, ranging from 10% (IHC) up to 26% (RT-PCR).
From another research group [93, 94], there are two more exhaustive studies that used an elevated number of detailed characterised histological FFPE samples and FNA (fine needle aspiration) cytological smears to accurately quantify the results. We recovered from these papers some qRT-PCR data that are shown in Table 2: (i) normal tissues, nodules, goitres, and thyroiditis are HMGA2 negative; (ii) follicular adenomas show very low positivity; (iii) follicular carcinomas, papillary carcinomas, and anaplastic carcinomas have very high levels of positivity. The consistency between the two types of analysed specimens, i.e., FFPE and FNA, is striking, even though they were obtained using very different sampling procedures. The authors concluded that because of the very high sensitivity methodologies and specificity “HMGA2 qRT-PCR can be used to distinguish benign from malignant thyroid tumors in both FFPE and FNA specimens”. Here, we want to specifically stress the HMGA2/FNA results because this procedure, being preoperative and directed towards gaining information on suspicious cases, could constitute real progress in clinical thyroid diagnosis.
To more reliably interpret the analytical results from molecular factors, many studies describe panels of more factors that should substantiate the final conclusion when considered together. Along this line of research is the paper by Prasad et al. [95] which analysed HMGA2 expression together with a group of 9 other factors in specimens from FFPE and FNA normal and tumour tissues by both IHC and qRT-PCR. The authors demonstrated that, according to the specificity and sensitivity, a test of the three factors HMGA2, SFN, and MRC2 has the potential to differentiate benign from malignant tumours in thyroid cancer. In Table 2 we summarised most of the results reported in the above studies for HMGA2 in thyroid specimens. Table 2 reveals similarities among the data obtained by the various research groups; we can conclude that, in the assessment of thyroid tissue samples, the evaluation of the expression of HMGA2 should be preferred to that of HMGA1 because there are much more data for the former in comparison with the latter. Additionally, among HMGA2 results, qRT-PCR appears to be more reliable than IHC. As a general conclusion, it is possible to confirm that, considering all of the HMGA2 data, a negative result should correspond to benign tissue, while a positive result very likely arises from a malignant carcinoma. This last sentence is also founded on a statistical χ2 test (Pearson) that we performed using all of the HMGA2 data in Table 2. We grouped together normal tissues, goitres, thyroiditis, nodules, and adenomas: Table 2A, a) and b) (154 total RT-PCR cases); we also grouped together all carcinomas: Table 2B, FTC (follicular thyroid carcinoma), PTC (papillary thyroid carcinoma), FVPTC (follicular variant papillary thyroid carcinoma), and ATC (anaplastic thyroid carcinoma), (c), (d), and (e) (289 total RT-PCR cases). The result of the test was highly significant: p < 0.0001. The same result was also obtained including the IHC data.
Brain cancerTop
Elevated levels of HMGA1 expression were detected in various types of brain tumours (astrocytoma, glioblastoma, and medulloblastoma) by IHC and qRT-PCR and were related to an advanced degree of neoplasia and poor prognosis [96-98]. HMGA1 over-expression has been related to several significant biological processes in brain tumours: i) proliferation as measured by Ki67/MIB1 (a known marker of proliferation) and cdc25A (a phosphatase that regulates cell cycle) [99], shown here by up-regulation at the promoter by HMGA1; ii) migration/invasion through MMP-9 (a matrix metallo-protease strongly associated with inflammation and cancer [100, 101], and VEGFA (a vascular endothelial growth factor which promotes neovascularisation and invasion and is linked to cancer-transforming pathways as found in glioblastoma) [102, 103]. Here, we introduce a digression that concerns other types of cancer (gastric and melanoma), but it is of interest because of the elevated invasiveness of brain tumours. The paper by Fernández et al. [103] reports that the factor named survivin enhances the activity of the β-catenin/Tcf/Lef complex and enhances the expression of many genes, including survivin itself (representing positive feedback loop) and VEGF. VEGF is then secreted into the extra-cellular compartment and angiogenesis and tumour growth are promoted. Consistently, in a study by Wang et al. [104] using 80 cases of brain glioma together with human glioma cell lines, by IHC, qRT-PCR, and techniques to silence/over-express survivin, it was demonstrated that VEGF is essential during glioma angiogenesis. However, survivin and VEGF are linked, in another tumour type OSCC (oral squamous cell carcinoma) to the other protein, i.e., HMGA2 because the stable expression of Lin28B (by suppressing let-7) increases the expression of genes such as VEGF, HMGA2, the EMT markers Snail and Twist, and survivin, promoting cell migration, invasion, colony formation, and proliferation [105] (Figure 9a).
Two papers dealing with HMGA2 expression and regulation have been recently published on malignant gliomas including glioblastoma multifome and anaplastic astrocytoma [106, 107]. The expression results are very similar to those found in HMGA1 studies. Indeed, both glioblastoma multiforme and anaplastic astrocytoma, analysed by IHC and qRT-PCR, show high levels of HMGA2 expression significantly associated with a shorter progression-free survival time of the patients. Similarly to HMGA1, proliferation and invasion are the biological effects of HMGA2 over-expression according to the proliferation marker Ki-67/MIB1 and to the over-expression of the metalloprotease MMP-2 that we previously reported to also be induced by HMGA1 in lung cancer [58]. Referring to other types of cancer, on the one hand, HMGA1 and HMGA2 are able to induce the metalloproteases MMP-2 and MMP-9 (Brain and Lung), but, on the other hand, another important factor, Sox-2 (Sex determining Y box 2 (Breast, Brain, and CRC) an inducer of pluripotent stem cells and invasiveness. Sox-2 activity is linked to the Wnt pathway and stem cells which we know are closely connected with the HMGA1/HMGA2 proteins, and its property to stimulate invasion is mediated by MMP-2 activity [20, 107, 108]. Furthermore, inhibition of the expression of HMGA2 represses Sox-2, tumorigenicity and cancer stem cell-like properties of anaplastic astrocytoma-derived cells. The inhibition of HMGA2 was achieved by the delivery of a vector-mediated let-7a miR repressor; we previously introduced let-7a in the Prostate section when discussing the paper by Schubert et al. [42] (Figure 9b).

In glioma cells, the expression of let-7a is also able to induce cell cycle arrest and apoptosis, and the inhibition of migration and invasion by repressing the PI3K/Akt and K-Ras/MAPK/ERK pathways [109]. Moreover, according to the paper by Mao et al. [110], along the same line of research, the increased expression of Lin28A in glioblastoma cells down-regulates let-7 miRs (b and g) increasing the expression of HMGA2 and the SNAI1 EMT gene accompanied by the enhanced aggressiveness and invasiveness of cells. We discussed the b form of let-7 miRs in the CRC section because of its relationship with IGF2BP2 and Lin28B, of which the former is positively induced by HMGA2 [31, 35]. In conclusion, Lin28A and B down-regulate let-7a and b to induce the HMGA2 expression that is associated with stem-like cell properties in various types of cancer (Figure 10a, b).

IGF2BP2, also known as IMP2, and its homologue IMP1 are deeply involved in brain biology because they regulate neuronal development together with HMGA proteins in the context of global chromatin structural organisation. Indeed, the differentiation program of neuronal precursor cells (NPCs) to give rise to neuronal and glial type cells requires both sets of factors (IMPs and HMGA), although not necessarily following the same order of events. According to Nishino et al. [111], IMP1 is highly expressed in progenitor neuronal stem cells where it stabilises a number of mRNAs that produce self-renewal stem cell factors such as HMGA2, but (interestingly) not HMGA1. In the adult neuronal stem cells, the binding of let-7 miR down-regulates IMP1 that could be re-expressed in cancer cells as a consequence of activation of the Wnt pathway. The reader is certainly going to notice that, here, we return to the beginning of this review (Breast section) where the Wnt/β-catenin pathway was introduced. Similarly, HMGA2 induced IMP2 at the early stages, giving NPCs neurogenic potential and, at the same time, inhibiting their astrocytic differentiation [112]. Consistently, selected glioblastoma tumour samples show a sub population of cancer stem cells (CSCs) expressing high IMP2 levels that assure self-renewal by supplying the energy requirements for cell growth through mitochondrial oxidative phosphorylation [113] (Figure 11a).
The huge amount of HMGA proteins present in cancer cells allowed for their primary identification by the chemical methods [1-4] used in the study of other, even more abundant nuclear proteins, i.e., histones, for which a definite chromatin structural function was already known. These high mass levels of HMGA proteins, accompanying histones and DNA suggested a global contribution by HMGA protein to the chromatin organisation besides being specifically localised factors for the regulation of gene expression [114, 115]. Recently, the point has been highlighted by an elegant and exhaustive work by Kishi et al. [116], where studying the neuronal precursor cells (NPCs) of the mouse neocortex demonstrated, by a careful digestion of selected fractions of chromatin, that HMGA proteins regulate the global chromatin state and establish an open structure able to mediate the neurogenic potential of early stages of NPCs. The authors suggest a similar or redundant function for HMGA1 and HMGA2 in promoting the self-renewal and proliferation of NPCs. In a recent review, Ozturk and colleagues [117] reported, in a compact form, a clear vision of the involvement of HMGA proteins in modulating, together with histones and transcription factors, chromatin structure and related gene expression.
The metastasis suppressor activity of RKIP, which was discussed in the Prostate section, can also be observed in glioma cells in which, as in previous cancers [22, 54, 55], it is able to inhibit cell invasion through the up-regulation of miR-98 in the context of the coordinated Lin28/let-7-miR-98/HMGA2 axis that results in the down-regulation of HMGA2 [118]. It should be mentioned that miR-98 over-expression mainly acts as an inhibitor of glioma cell invasion rather than as an inhibitor of cell proliferation. Another HMGA2 suppressor in glioblastoma is miR-142-3p [119] that can be down-regulated by IL-6, in turn, which is known to induce HMGA2 in breast cancer by the Stat3/Lin28/let-7 pathway. The over-expressed HMGA2 is able to enhance Sox-2 expression that is related to cell stemness. Moreover, miR-142-3p is also involved in another pathway related to HMGA2 and proliferation, i.e. the Wnt/β-catenin pathway. This miR binds to APC (Adenomatous Polyposis Coli) RNA and regulates the level of β-catenin during lung development, balancing the proliferation or differentiation of embryonic mesenchymal cells [120]. Stat3 is deeply involved in brain tumours because its hyper-phosphorylation, dimerisation, and nuclear translocation is induced upstream by a variety of cytokines (such as IL-6 and other interleukins) and growth factors (such as EGF, PDGF, HGF, and FGF), which are responsible for the expression of factors such as cyclin D1, Bcl-2, c-Myc, VEGF, MMP-2, and MMP-9. All of these factors are involved in glioblastoma in which the Stat3 pathway is further potentiated because of the absence of physiological regulatory inhibitors [121]. Therefore, inhibitor molecules have been developed (for example WP1066 and STX-0119) to target Stat3; these molecules have been used in cells derived from glioblastoma and showing cancer stem-like properties similar to those present in tumours, and are assumed to be responsible for recurrence and therapeutic resistance [122, 123].
A variety of miRs are involved in brain tumours as onco-inducers or onco-suppressors whose dysregulation can occur as an over-expression or under-expression [124, 125]. miR-10b, miR-21, miR-125b, and miR-221 are up-regulated in thyroid cancer by HMGA1, while miR-34a is down-regulated, exhibiting the same behaviour in glioblastoma, although they are not explicitly related to HMGA1/A2 proteins in these studies. The importance of miR-34a has been previously noted; in glioblastoma stem cells, it inhibits invasion/migration, proliferation, cell cycle progression, stemness, and tumorigenicity, but (consistently) promotes apoptosis [126]. This is accomplished by relationships that miR-34a (and its family members b and c) establishes with a myriad of factors, only a few of which will be cited here because they are directly or indirectly related to HMGA1/A2 proteins. A more complete list of these factors has been reported by Rokavec and colleagues [127]. The positive induction of miR-34a by p53 is definitively demonstrated in various types of tumours, including gliomas; however, the effect on miR-34a is reduced in glioblastomas carrying mutant p53. When p53 is functioning and miR-34a is expressed, the results are: 1, cell cycle, hampered (by repressing c-Myc and HMGA2); 2, invasion, inhibited (by repressing IL-6R); 3, apoptosis, induced (by Bcl-2 repression); 4, EMT, inhibited (by SNAIL down-regulation); 5, Wnt signalling, stemness, EMT, and metastasis, all inhibited (by down-regulating Sox-2, Lef1, and the Notch pathway); and 6, differentiation, stimulated (by Lin28A repression). Finally, it is worthwhile to mention that miR-34a inhibition increases survivin expression supporting both proliferation and invasion, but its expression (interestingly) decreases the resistance of cisplatin-resistant gastric cancer cells by modulating the PI3K/Akt pathway [128, 129]. Figure 11b shows miR-34a relationships in glioblastoma.

DiscussionTop
Our purpose was to review the expression of HMGA1/A2 proteins in the context of a number of oncogenic transforming pathways. Inspection of the various pathways we presented allows us to infer that some pathways induce the expression of both HMGA1 and HMGA2: Ras/Raf (Figures 3, 6, and 7), Wnt/β-catenin (Figures 2 and 3), and TGF-β (Figures 3 and 5). Conversely, it seems that the two pathways PI3K/Akt and IL-6/Stat3 are only involved, at least directly, in the expression of one of the two: the former in HMGA1 (Figures 4, 6, 7, and 8), and the latter in HMGA2 (Figure 2). However, this summary is rather schematic and simplistic because cross-linked inductions that promote the expression of one or the other protein are possible. For example, HMGA1 can induce Stat3 (Figure 2) and consequently HMGA2, together with the Ras and Wnt/β-catenin pathways. On the other hand, PI3K/Akt is responsible for HMGA1 expression (Figure 6). Indeed, it emerges that HMGA1/A2 are the result of various pathways and simultaneously constitute self-inducing factors and factors that induce other pathways, establishing a series of perverse loops by which the tumour enhances its capability to grow, invade, and achieve the extreme point of spreading metastases. By combining the information summarised in the figures, other positive cooperating loops can be deduced that are all aimed at an unrestrainable and complete tumour transformation.
In the complex system of interlinked transforming pathways being discussed here, the regulatory organisation of other factors is superimposed and can promote or hinder tumour development and invasion associated with HMGA1/A2 expression. Lin28/let-7 is the main regulatory axis that regulates HMGA1/A2 expression as reported above and further supported by data of the literature [130-133]. The let-7 miR family is the most well-known inhibitory system, but many other regulatory miRs of the HMGA1/A2 proteins have been reported, some of which we previously cited. The regulation of the expression of the HMGA1/A2 proteins brings up a question about their redundancy. In other words, can one protein completely substitute the other or, in terms of its action, can one protein do without the other? A similar question was asked by Shah et al. [26]. They hypothesised a possible functional redundancy of the HMGA1 and HMGA2 proteins. Table 1 of this review, the other data throughout the text, and the Figures clearly show a functional redundancy of the two proteins as a final result, i.e., transformation, cell cycle, stemness, invasiveness, and EMT. However, in most cases, the function of only one protein has been studied and the other remains less clear. From the point of view of the cell economy, it is unreasonable to use different cellular machinery to produce proteins that have the same overlapping functions. It is likely that HMGA1 and HMGA2 could be present from time to time according to the fine tuning of the regulatory axis Lin28/let-7 or others. The A and B forms of Lin28 regulate the expression of HMGA1/A2 by repressing the let-7 family of miRs. Lin28A and Lin28B exert their let-7 regulation by a distinct mechanism, and they could consequently have different effects on the thirteen members of the let-7 family. However, Lin28A and B could also be considered functionally redundant proteins that block let-7miRs, as in body development [134]. It has been reported that Lin28A and Lin28B are differentially expressed in tumours such as breast and colon tumours. However, their over-expression is consistently detected in less than 50% of the analysed tumour samples. Interestingly, tumours in which Lin28A is expressed at high levels show low levels of Lin28B and vice versa. Taken together, this means that let-7 may also be regulated by different mechanism. To further increase the complexity of Lin28/let-7/HMGA regulation, it is interesting to note that reverse regulation of Lin28 expression by the HMGA1 protein has also been reported as demonstrated by the fact that the knockdown of HMGA1 represses Lin28 [26]. Finally, HMGA1/A2 probably work together with other factors (not necessarily the same factors) to form multi-functional complexes by establishing protein-protein interactions using the regions of the proteins that are non-homologous (approximately 50%) (Figure 1) [135, 136].
The selective regulation of the HMGA1/A2 proteins could be also possible through the different forms of let-7 family factors. The data of the text and Figures (though numerically limited) show that HMGA1 is regulated by the let-7b form and by miR-296, whereas HMGA2 is regulated by the a, b, and g let-7 forms (Figures 4, 6, 7) in different types of cancer: lung, prostate, hepatocellular carcinoma, ovarian, and glioma. Among the miR forns, a clear specificity has been reported for the c form that is unable to repress HMGA1 [42]. Concerning this differential regulation, it is worth mentioning the study of Shlapobersky et al. [137], who studied the expression of HMGA2 in human cytomegalovirus-infected foreskin fibroblasts. An efficient replication of the virus requires the down-regulation of both HMGA2 and cyclin A, the latter being under the control of the former [138]. Interestingly, there was much less of a reduction in HMGA1. Among the other miRs that are involved in the positive or negative regulation of the HMGA1/A2 proteins, we emphasised miR-34a because it is coupled with p53, which is one of most studied tumour suppressors and we are interested in understanding the action of miR-34a and p53 on HMGA1/A2 expression. p53 interacts with and stimulates miR-34a that is then able to down-regulate a series of factors (including HMGA1/A2), pathways, and tumour properties, as shown in figures 5, 8, and 11B with the last highlighting the adverse consequences that active p53/miR-34a can have on tumour development.
A tentative conclusion regarding the above questions could be that the effects of HMGA1/A2 could be redundant through a differential regulation of their expression and, when they are expressed together, according to the observed inter-linked pathways, a more transformed system is achieved. However, another question remains. Which occurs first? In other words, is there any priority in the expression of one protein over the other?
It is interesting to examine some of the data of the three previously cited studies in detail [16, 36, 116]. The paper by Wend et al. [16], in addition to the study of MDA-MB-231 cells, as discussed in the Breast section, also reports investigations in other cell lines: 1) the non-triple negative breast cancer cell line MCF-7 has low Wnt/β-catenin activity and expresses very low levels of HMGA1/A2 proteins that are barely detectable; 2) the triple-negative breast cancer cell lines MDA-MD-231, BTL10, and MA-11 express high levels of both proteins, and have high Wnt/β-catenin activity; however MDA-MD-231, BTL10, and MA-11 exhibit predominant increases of the expression of HMGA2 in comparison with HMGA1 (approximately 24, 100, and 17 times, respectively) when normalised to MCF-7 cells; and 3) the breast cancer cell line MCF10b, with an active Wnt/β-catenin pathway shows almost undetectable mRNA expression levels of HMGA1, whereas HMGA2 mRNA is expressed at very high levels (both normalized to human MCF-7). The data from these cell lines, in addition to confirming that the Wnt/β-catenin pathway is responsible for the expression of the proteins in breast cancer, demonstrate that: (i) the existence of cancer cells expressing both proteins is possible and (ii) the expression of HMGA2 essentially without HMGA1 is possible. It is not clear whether the reverse is also possible, i.e. HMGA1 without HMGA2. There are much data indicating that this is the most frequent situation. For example, the paper by Li et al. [36], which reported a study on rhabdomyosarcoma in which the highly expressed proteins HMGA2/IGF2BP2 were shown to induce tumour survival and growth, also studied a group of cancer/immortalised cell lines in which the expression of both HMGA1 and HMGA2 proteins was investigated. It was found that HMGA1 is ubiquitous and constantly expressed in the cell lines HEK-213 (human embryonic kidney), MDA-453 (breast cancer), MCF-7 (breast cancer), RD (rhabdomyosarcoma), and TE617 (rhabdomyosarcoma). Conversely, HMGA2 was only expressed in RD and TE617 cells, constituting a kind of completeness for full cell transformation. The authors further emphasised these findings by stating that HMGA1 mRNA is ubiquitously expressed in various normal and malignant tissues. Unlike HMGA2 mRNA, HMGA1 mRNA does not show differential expression between various normal tissues and cancer cells. However, this sentence cannot be assumed to be a general rule. Indeed, in most papers on tumour tissues, the nuclear positivity was determined by comparison with a normal counterpart tissue, which includes the tissue surrounding the tumour or a tissue sample from a related healthy organ. Not only was the expression observed but also its differential increase with the progress of transformation [34, 58, 71, 83, 84, 89, 96, 98, 139]. When comparing HMGA1 expression with HMGA2 expression, the previously discussed study of ovarian cancer by Mahajan et al. [70] is significant. In the six different types of ovarian tumours, HMGA1 expression is roughly constant (measured as the number of positive cells and staining intensity), whereas HMGA2 expression is variable. Interestingly, the highest levels are present in the most transformed tumour, i.e., the high-grade papillary serous carcinoma (HG-PSC). Considering these results together with results from the cell lines discussed above, it is possible to conclude that the expression of HMGA2 provides an additional contribution to the transforming activity by HMGA1, followed by a mutual increase in their action that results in a very high efficiency level of the system. This highly related cooperation between the two proteins was first shown in a cell system in which a profound genetic alteration was induced by retrovirus infection of rat thyroid differentiated cells, which induced a contemporaneous elevation of the expression of HMGA1a, A1b, and A2 [1-4]. The transfection of an antisense construct for HMGA2 in these cells, at the same time as virus-infected, not only caused a strong reduction of the HMGA2 protein but also of the HMGA1a/1b proteins and the cells were no longer neoplastically transformed [140].
This review discusses the HMGA1/A2 proteins in cancer, but there are many other roles of the HMGA1/A2 proteins that are of elevated interest, such as their role in development. It has been shown by in vivo experiments using HMGA1- and Hmga2-null mice that the two proteins independently regulate different metabolic points in development, i.e., are not redundant. We do not discuss these studies in detail, but there are some studies that are closely associated with the HMGA1/A2 cooperation that we will discuss here. In the paper by Kishi et al. [116], the cooperation between HMGA1 and HMGA2 in the brain and in normal development is clearly demonstrated. Indeed, the presence of HMGA proteins in the chromatin allows for augmented extraction (NaCl) of the histones H2A/H2B from the nucleosomes. If only HMGA1 is present, 17% of H2A/H2B is extracted, whereas if only HMGA2 is present, 24% of H2A/H2B is extracted. However, if both HMGA1 and HMGA2 are present on the chromatin, creating an even more open structure, 44% of H2A/H2B is extracted. These results give rise to two conclusions: (i) to open the compact chromatin structure, each protein can work independently from the other and not necessarily on the same nucleosomes; (ii) to induce the differentiation of neocortical NPCs and increase the number of newborn neurons, the expression of both HMGA1/A2 proteins is much more efficient than the expression of only one of them. Since the pioneering work by K Chada’s group [141], the integrity of the HMGA2 gene is fundamental for normal development in the mouse because its inactivation generates a pygmy phenotype. As recently demonstrated by Federico et al. [142], double knock-out of Hmga1 and Hmga2 cooperatively results in an even more dramatic effect: a further decrease in the growth rate and a striking reduction in body size are observed, and the phenotype of the generated pygmy mice has been named super pygmy.
Through the description of HMGA1/A2 data, we showed several links (expression and action) with other factors. The story of some of them is strictly associated with that of HMGA1/A2, such as Sox-2 and cMyc, for which questions about expression hierarchy and redundancy could be brought up again. Despite the fact that Sox-2 is an HMG protein belonging to the HMGB sub-class, its expression and functions are intertwined with those of HMGA1/A2. Indeed, Figures 2, 9, and 11b show that HMGA1/A2 in breast and brain tumours preside over Sox-2 expression, and HMGA2 inhibition down-regulates Sox-2. Paralleling the cellular features induced by HMGA1/A2, Sox-2 expression promotes the reprogramming of somatic cells into stem-like phenotype, including migration, invasiveness, and metastasis. Consistently, Sox-2 expression increases the expression of mesenchymal markers; silencing of Sox-2 drastically down-regulates cMyc, Lef1/Ctf1, SNAI1/2, ZEB1/2, Twist (all EMT inducers), and a group of mesenchymal cell markers, while the expression of many epithelial markers are strongly activated [143, 144]. It is logical that Sox-2 is integrated into the Wnt/β-catenin pathway together with HMGA1/A2 because it shares the same Lin28B/let-7 regulatory axis.
Pathways that are already active due to various causes (extracellular stimuli and mutations), such as K-Ras, Wnt/β-catenin, PI3K/Akt, and IL-6/Stat3, can induce the expression of cMyc according to Figures 2, 6, 7, and 11b and related References. Because of the established presence of many regulating loops, several scenarios could be possible. For example, if an active K-Ras induces the expression of Lin28B, cMyc is expressed because the let-7 family is repressed [145]. Together with cMyc, HMGA2 is also expressed. Conversely, if the let-7 family is active (in particular the let-7d form), there is direct inhibition of the Ras family (K-Ras, H-Ras, and N-Ras) as well as cMyc and HMGA2. Interestingly, let-7d does not inhibit HMGA1. Moreover, it has been reported that the let-7 family negatively regulates HMGA1, HMGA2, cMyc, N-Ras, and IL-6. However, the expression of cMyc induces Lin28 followed by the inhibition of let-7 maturation [146-148].
Different regulatory loops could be functioning that are essentially centred on the triad cMyc/Lin28/let-7: The branches linking the factors form a closed loop. If the loop has self-supporting activity, as we provided evidence for in the text, then the tumour progresses. If stimulatory and inhibitory branches are both functioning and have equivalent quantitative effects, then nothing changes. Because a tumour has an initiating point (not always definite) and an extreme final point (metastasis), the branches that support proliferation and invasion must prevail over the others of the loop.
In the description of the pathways that activate the expression of the HMGA1/A2 proteins, we reported other factors that are also over-expressed and related to the invasive properties of the tumour. This process results in the EMT in which three pairs of factors play major roles together with HMGA1/A2, making up a cooperating group of factors, SNAI1/2, ZEB1/2, TWIST1/2, and HMGA1/A2, which are tightly linked in EMT, and are all involved in E-cadherin down-regulation. Some of the reported Figures show that the main cancer-active pathways induce more or less the complete set of these factors and HMGA1/A2 proteins. In breast cancer (Figure 2) [18], Oncostatin (OSM and IL-6), promotes the expression of Lin28B that induces ZEB1 (and HMGA2) by repressing miR-200 and let-7 family. In glioblastoma, a feed-back loop between miR-200 and ZEB1 in which ZEB1 inhibits miR-200 is observed. If miR-200 inhibition prevails, tumour initiation by Sox-2 occurs, followed by invasion (EMT) and chemoresistance [149]. Similarly, in undifferentiated endometrial carcinomas [150], ZEB1 over-expression is linked to miR-200 and E-cadherin down-regulation. The TGF-β pathway induces ZEB1/2 that down-regulates miR-200, which in turn (if expressed) is able to repress ZEB1/2 and TGF-β (again the same feed-back loop). Of course, a prevailing active TGF-β should induce ZEB1/2 followed by tumour progression and EMT stabilisation [151]. Almost all of the factors are also induced by the TGF-β/Smads/HMGA2 system: in mammary epithelial cells, SNAI1/2, ZEB1/2, and TWIST1 [152, 153]; in lung tissue, ZEB1/2 and HMGA2 [154]. Moreover, for deep invasion and metastasis, high levels of expression of HMGA2, TWIST1, and ZEB1 have been observed in melanoma [155], and high levels of expression of HMGA2, SNAI2, and ZEB1 have been observed in myometrial carcinoma [77].
Together with the two EMT-inducer pathways discussed above (TGF-β and IL-6/Stat3), there is also the obvious involvement of the other three important pathways, i.e. Wnt/β-catenin, K-Ras/B-Raf, and PI3K/Akt, as shown in the reported Figures. For instance, K-Ras/B-Raf (Figure 7) indicates, for active mutant K-Ras/B-Raf, a huge amount of induced factors that participate in the process under discussion. In conclusion, there are intricate interactions that link the pairs of reported factors and other factors in stemness, invasion, and chemoresistance. Among them, HMGA2 and ZEB1 seem to prevail, as demonstrated by their widespread expression and increased concentration at the tumour invasive front. HMGA2 rather than HMGA1 appears to be more associated with the other EMT factors which could represent an additional contribution of HMGA2 to already cancerous cells expressing HMGA1. As discussed for the HMGA1/A2 proteins, and for the three other pairs of factors, it could be a possible question of redundancy. In this case, the absence of overlapping activity could be avoided by selective temporal and spatial protein expression. Indeed, the question has already been raised for TWIST1/2 by Franco et al. [156] because the two proteins have 66% total sequence identity and up to 98% sequence identity in the HLH functional region. Various hypotheses have been formulated to exclude an overlapping action including different regulation of the genes, different post-translational modifications, and different interactions with other transcription factors. In any case, the full extent to which these proteins diverge in function remains to be seen [156].
ConclusionTop
The whole set of cooperating HMGA and other factors: i) if expressed, regulate increasing invasion and aggressiveness in tumour cells; ii) when induced in somatic cells, cause phenotypic reprogramming towards stem-like cells; iii) the same factors could regulate the following re-differentiation. The presence of HMGA2 appears to drive the system to the highest status of functional activity. In other words, there is self-renewal and proliferation activity that is higher than that shown in other cancer cells of the same tissue. Another point of relevance is EMT-sustained invasiveness in which HMGA2 appears to represent a pivotal point because of its links with the other EMT-involved factors. There is no evidence to suggest a similar redundant function by HMGA1. The resistance to drugs and radiations that sub-groups of cancer cells show is suggested to be caused by special stem cells responsible for relapse that express high levels of the HMGA1/A2 proteins. The resistance should result in the inhibition of apoptosis that we associated with Bcl-2, miR-34a, and HMGA1/A2, although other mechanisms are possible such as increased activity of DNA-repair mechanisms due to the proteins under discussion.
Acknowledgment
Acknowledgments are due to Trieste Proteine Ricerche for office and graphical assistance.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
HMGA: High mobility group A; HMGA1a: High mobility group A1a; HMGA1b: High mobility group A1b; HMGA2: High mobility group A2; ESCs, Embryonic stem cells; ASCs: Adult stem cells; CSCs: Cancer stem cells; EMT: Epithelial-mesenchymal transition; TNBC: Triple negative breast cancer; miR: Micro-RNA; IHC: Immunohistochemistry; OSM: Oncostatin M; JAK: Janus kinase; IL-6: Interleukin-6; RTKs: Receptor tyrosine kinases; CRC: Colorectal cancer; TGFβRII: TGF-β receptor II; IGF-2BP2: Insulin-like growth factor-2 m-RNA binding protein 2; SP1: Specific protein 1; AP1: Activator protein 1; HGF: Hepatocyte growth factor; RKIP: Raf kinase inhibitor protein; MMP-2: Matrix metallo-protease-2; MMP-9: Matrix metallo-protease-9; TTF-1: Thyroid transcription factor-1; HG-PSOC: High-grade papillary serous ovarian carcinoma; FFPE: Formalin-fixed paraffin-embedded; FNA: Fine needle aspiration; FTC: Follicular thyroid carcinoma; PTC: Papillary thyroid carcinoma; FVPTC: Follicular variant papillary thyroid carcinoma; OSCC: Oral squamous cell carcinoma; VEGFA: Vascular endothelial growth factor A; Sox-2: Sex determining Y box 2; NPC: Neuronal precursor cells; APC: Adenomatous polyposis coli
ReferencesTop
[1]Lund T, Holtlund J, Fredriksen M, Laland SG. On the presence of two new high mobility group-like proteins in HeLa S3 cells. FEBS Lett. 1983; 152(2):163–167.Article Pubmed
[2]Goodwin GH, Cockerill PN, Kellam S, Wright CA. Fractionation by high-performance liquid chromatography of low-molecular-mass high--mobility-group (HMG) chromosomal proteins present in proliferation rat cells and an investigation of the HMG proteins present in virus transformed cells. Eur J Biochem. 1985; 149(1):47–51.Article Pubmed
[3]Giancotti V, Berlingieri MT, Di Fiore PP, Fusco A, Vecchio G, et al. Changes in nuclear proteins on transformation of rat epithelial thyroid cells by a murine sarcoma retrovirus. Cancer Res. 1985; 45(12 pt 1):6051–6057.Article Pubmed
[4]Giancotti V, Pani B, D’Andrea P, Berlingieri MT, Di Fiore PP, et al. Elevated levels of a specific class of nuclear phosphoproteins in cells transformed with v-ras and v-mos oncogenes and by co-transfection with c-myc and polyoma middle T genes. EMBO J. 1987; 6(7):1981–1987.Article Pubmed
[5]Johnson KR, Lehn DA, Elton TS, Barr PJ, Reeves R. Complete murine cDNA sequence, genomic structure, and tissue expression of the high mobility group protein HMG-I(Y). J Biol Chem. 1988; 263(34):18338–18342.Article Pubmed
[6]Manfioletti G, Giancotti V, Bandiera A, Buratti E, Sautiere P, et al. cDNA cloning of the HMGI-C phosphoprotein, a nuclear protein associated with neoplastic and undifferentiated phenotypes. Nucleic Acids Res. 1991; 19(24):6793–6797.Article Pubmed
[7]Bustin M. Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem Sci. 2001; 26(3):152–153.Article Pubmed
[8]Johnson KR, Lehn DA, Reeves R. Alternative processing of mRNAs encoding mammalian chromosomal high-mobility-group proteins HMG-I and HMG-Y. Mol Cell Biol. 1989; 9(5):2114–2123.Article Pubmed
[9]Friedmann KR, Holth L, Zoghbi HY, Reeves R. Organisation, inducible expression and chromosomal localisation of the human HMG-I(Y) non-histone protein gene. Nucleic Acids Res. 1993; 21(18):4259–4267.Article Pubmed
[10]Patel U.A, Bandiera A, Manfioletti G, Giancotti V, Chau KY, et al. Expression and cDNA cloning of human HMGI-C phosphoprotein. Biochem Biophys Res Commun. 1994; 201(1):63–70.Article Pubmed
[11]Chau KY, Patel UA, Lee K, Lamm HY, Crane-Robinson C. The gene for the human architectural transcription factor HMGI-C consists of five exons each coding for a distinct functional element. Nucleic Acids Res. 1995; 23(21):4262–4266.Article Pubmed
[12]Dzobo K, Vogelsang M, Parker MI. Wnt/β-catenin and MEK/ERK signaling are required for fibroblast-derived extracellular matrix-mediated endoderm differentiation of embryonic stem cells. Stem Cell Rev. 2015; 11(5), 761–773.Article Pubmed
[13]Miki T, Yasuda S, Kahn M. Wnt/β-catenin signaling in embryonic stem cell self-renewal and somatic cell reprogramming. Stem Cell Rev. 2011; 7(4):836–846.Article Pubmed
[14]Neth P, Ries C, Karow M, Egea V, Ilner M, et al. The Wnt signaling transduction pathway in stem cells and cancer cells: Influence on cellular invasion. Stem Cell Rev. 2007; 3(1):18–29.Pubmed
[15]Van Camp JK, Beckers S, Zegers D, Van Hul W. Wnt signaling and the control of human stem cell fate. Stem Cell Rev. 2014; 10(2):207–229.Article Pubmed
[16]Wend P, Runke S, Wend K, Anchordo B, Yesayan, et al. WNT10B/β-catenin signaling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol Med. 2013; 5(2):264–279.Article Pubmed
[17]Cai WY, Wei TZ, Luo QC, Wu QW, Liu QF, et al. The Wnt/β-catenin pathway represses let-7 microRNAs expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J Cell Sci. 2013; 126(Pt 13):2877–2889.Article Pubmed
[18]Guo L, Chen C, Shi M, Wang F, Chen X, et al. Stat3-coordinated Lin-28-let-7-HMGA2 and MiR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene. 2013; 32(45):5272–5282.Article Pubmed
[19]Shah SN, Cope L, Poh W, Belton A, Roy S, et al. HMGA1: A master regulator of tumor progression in triple-negative breast cancer cells. PLoS One. 2013; 8(5):e63419.Article Pubmed
[20]Di Cello F, Shin J, Harbom K, Brayton C. Knockdown of HMGA1 inhibits human breast cancer cell growth and metastasis in immunodeficient mice. Biochem Biophys Res Commun. 2013; 434(1):70–74.Article Pubmed
[21]Pegoraro S, Ros G, Oiazza S, Sommaggio R, Ciani Y, et al. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013; 4(8):1293–1308.Article Pubmed
[22]Dangi-Garimella S, Yun Y, Eve EM, Newman M, Erkeland SJ, et al. Raf kinase inhibitor protein suppresses a metastasis signaling cascade involving LIN28 and let-7. EMBO J. 2009; 28(4):347–358.Article Pubmed
[23]Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle. 2010; 9(12):2363–2374.Article Pubmed
[24]Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005; 17(5):548–558.Article Pubmed
[25]Tan EJ, Thuault S, Caja L, Carletti T, Heldin CH, et al. Regulation of transcription factor Twist expression by DNA architectural protein high mobility group A2 during epithelial-to-mesenchymal transition. J Biol Chem. 2012; 287(10), 7134–7145.Article Pubmed
[26]Shah SN, Kerr, Cope L, Zambidis E, Liu C, et al. HMGA1 reprograms somatic cells into pluripotent stem cells by inducing stem cell transcriptional networks. PLoS One. 2012; 7(11):e48533.Article Pubmed
[27]Hillion J, Dhara S, Sumter T, Mukherjee M, Di Cello F, et al. The high-mobility group A1a/signal transducer and activator of transcription-3 axis: An Achilles heel for hematopoietic malignancies? Cancer Res. 2008; 68(24):10121–10127.Article Pubmed
[28]Huang X, Meng B, Iqbal J, Ding BB, Perry A-M, et al. Activation of the STAT3 signaling pathway is associated with poor survival in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol. 2013; 31(36): 4520–4528.Article Pubmed
[29]Shibata S, Murushima H, Asakura T, Matsuura T, Eda H, et al. Three-dimensional culture using a radial flow bioreactor induces matrix metalloprotease 7-mediated EMT-like process in tumor cells via TGFbeta1/Smad pathway. Int J Oncol. 2009; 34(5):1433–1448.Article Pubmed
[30]Treff NR, Pouchnik D, Dement GA, Britt R, Reeves R. High-mobility group A1a protein regulates Ras/ERK signaling in MCF-7 human breast cancer cells. Oncogene. 2004; 23(3):777–785.Article Pubmed
[31]Morishita A, Raza Zaidi M, Mitoro A, Sankarasharma D, Szabolcs M, et al. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013; 73(14):4289–4299.Article Pubmed
[32]Alajez NM, Shi W, Wong D, Lenarduzzi M, Waldron J. Lin28b promotes head and neck cancer progression via modulation of the insulin-like growth factor survival pathway. Oncotarget. 2012; 3(12):1641–1652.Article Pubmed
[33]Zha L, Zhang J, Tang W, Zhang N, He, M, et al. HMGA2 elicits EMT by activating the Wnt/β-catenin pathway in gastric cancer. Dig Dis Sci. 2013; 58(3):724–733.Article Pubmed
[34]Rizzi C, Cataldi P, Iop A, Isola M, Sgarra R, et al. The expression of the high-mobility group A2 protein in colorectal cancer and surrounding fibroblasts is linked to tumor invasiveness. Human Pathol. 2013; 44(1):122–132.Article Pubmed
[35]Brants JR, Ayoubi TA, Chada K, Marchal K, Van de Ven WJ, et al. Differential regulation of insulin-like growth factor II mRNA-binding protein genes by architectural transcription factor HMGA2. FEBS Lett. 2004; 569(1–3):277–283.Article Pubmed
[36]Li Z, Zhang Y, Ramanujan K. Oncogenic NRAS, required for pathogenesis of embryonic rhabdomyosarcoma, relies upon the HMGA2-IGF2BP2 pathway. Cancer Res. 2013; 73(10):3041–3050.Article Pubmed
[37]Raskin L, Fullen DL, Giordano TJ, Thomas DG, Frohm ML, et al. Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J Invest Dermatol. 2013; 133(11):2585–2592.Article Pubmed
[38]Cleynen I, Huysmans C, Sasazuki T, Shirasawa S, Van de Ven W, et al. Transcriptional control of the human High Mobility Group A1 gene: basal and oncogenic Ras-regulated expression. Cancer Res. 2007; 67(10):4620–4629.Article Pubmed
[39]Bush BM, Brock AT, Deng JA, Nelson RA, Sumter TF. The Wnt/β-catenin/T-cells factor 4 pathway up-regulated high-mobility group A1 expression in colon cancer. Cell Biochem Funct. 2013; 31(3):228–236.Article Pubmed
[40]Belton A, Gabrovsky A, Bae YK, Reeves R, Iacobuzio-Donahue C, et al. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS One. 2012; 7(1):e30034.Article Pubmed
[41]Zong Y, Huang J, Sankarasharma D, Morikawa T, Fukayama M, et al. Stromal epigenetic dysregulation is sufficient to initiate mouse prostate cancer via paracrine Wnt signalling. Proc Natl Acad Sci USA. 2012; 109(50):3395–3404.Article Pubmed
[42]Schubert M, Spahn M, Kneit S, Scholz CJ, Jonlau S. Distinct microRNA expression profile in prostate cancer patients with early clinical failure and the impact of let-7 as prognostic marker in high-risk prostate cancer. PLoS One. 2013; 8(6):e65064.Article Pubmed
[43]Wei JJ, Peng Y, Shi G, Olca B, Yang X, et al. Regulation of HMGA1 expression by microRNA-296 affects prostate cancer growth and invasion. Clin Cancer Res. 2011; 17(6):1297–1305.Article Pubmed
[44]Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog. 2011; 10:20.Article Pubmed
[45]Jackson BL, Grabowska A, Ratan HL, MicroRNA in prostate cancer: Functional importance and potential as circulating biomarkers. BMC Cancer. 2014; 14:930.Article Pubmed
[46]Tacheuki I, Takaha N, Nakamura T, Hongo F, Mikami K, et al. High mobility group protein AT-hook 1 (HMGA1) is associated with the development of androgen independence in prostate cancer cells. Prostate. 2012; 72(10):1124–1132.Article Pubmed
[47]Wang S, Sun YM. The IL-6/JAK/STAT3 pathway: Potential therapeutic strategies in treating colorectal cancer (Review). Int J Oncol. 2014; 44(4):1032–1040.Article Pubmed
[48]Bishop JL, Thaper D, Zoubeidi A. The multifaceted roles of STAT3 signaling in the progression of prostate cancer. Cancers (Basel). 2014; 6(2):829–859.Article Pubmed
[49]Cho KH, Jeong KJ, Shin S.C, Kang J, Park CG, et al. STAT3 mediates TGF-β1-induced TWIST1 expression and prostate cancer invasion. Cancer Lett 2013; 336(1):167–173.Article Pubmed
[50]Zu X, Zhong J, Tan J, Tan L, Yang D, et al. TGF-β1 induces HMGA1 expression in human breast cancer cells: Implication of the involvement of HMGA1 in TGF-β signaling. Int J Mol Med. 2015; 35(3):693–701.Article Pubmed
[51]Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, et al. IL-R6/STAT3/miR-34a feed-back loop promotes EMT-mediates colorectal cancer invasion and metastasis. J Clin Invest. 2014; 124(4):1853–1867.Article Pubmed
[52]Jansson MD, Lund HA. MicroRNA and cancer. Mol Oncol. 2012; 6(6):590–610.Article Pubmed
[53]Ji Q, Hao X, Meng Y, Zhang M, Desano J, et al. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumourspheres. BMC Cancer. 2008; 8:266.Article Pubmed
[54]Yousuf S, Duan M, Moen EL, Cross-Knorr S, Brilliant K, et al. Raf kinase inhibitor protein (RKIP) blocks signal transducer and activator of transcription 3 (STAT3) activation in breast and prostate cancer. PLoS One. 2014; 9(3):e92478.Article Pubmed
[55]Yun J, Frankenberger CA, Kuo WL, Boelens MC, Eves EM, et al. Signalling pathway for RKIP and Let-7 regulates and predicts metastatic breast cancer. EMBO J. 2011; 30(21):4500–4514.Article Pubmed
[56]Sun M, Gomes S, Chen P, Frankenberger CA, Sankarasharma D, et al. RKIP and HMGA2 regulate breast tumor survival and metastasis through lysyl oxidase and syndecan-2. Oncogene. 2014; 33(27):3528–3537.Article Pubmed
[57]Di Cello F, Hillion J, Hristov A, Wood LJ, Mukherjee M, et al. HMGA2 participates in transformation in human lung cancer. Mol Cancer Res. 2008; 6(5):743–750.Article Pubmed
[58]Hillion J, Wood LJ, Mukherjee M, Bhattacharya R, Di Cello F, et al. Upregulation of MMP-2 by HMGA1 promotes transformation in undifferentiated, large-cell lung cancer. Mol Cancer Res. 2009; 7(11):1803–1812. Article Pubmed
[59]Kumar MS, Erkeland SJ, Pester R, Chen CY, Ebert MS, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008; 105(10):3903–3908.Article Pubmed
[60]Meyer B, Loeschke S, Schultze A, Weigel T, Sandkamp M, et al. HMGA2 overexpression in non-small cell lung cancer. Mol Carcinog. 2007; 46(7):503–511.Article Pubmed
[61]Wang YY, Ren T, Cai YY, He XY, MicroRNA let-7a inhibits the proliferation and invasion of nonsmall cell lung cancer cell lines 95D by regulating K-Ras and HMGA2 gene. Cancer Biother Radiopharm. 2013; 28(2):131–137.Article Pubmed
[62]Chen KJ, Hou Y, Wang K, Li J, Xia Y, et al. Reexpression of let-7g microRNA inhibits the proliferation and migration via K-Ras/HMGA2/Snail axis in hepatocellular carcinoma. Biomed Res Int. 2014; 742417.Article Pubmed
[63]Harada D, Takigawa N, Kiura K. The role of STAt3 in non-small cell lung cancer. Cancers (Basel). 2014; 6(2):708–722.Article Pubmed
[64]Rice SJ, Lai SC, Wood LW, Helsley KL, Runkle EA, et al. MicroRNA-33a mediates the regulation of high mobility group AT-hook 2 gene (HMGA2) by thyroid transcription factor 1 (TTF-1/NKX2-1). J Biol Chem. 2013; 288(23):16348–16360.Article Pubmed
[65]Scrima M. De Marco C, Fabiani F, Franco R, Pirozzi G, et al. Signaling networks associated with AKT activation in non-small cell lung cancer (NSCLC): New insights on the role of phosphatydil-inositol-3 kinase. PLoS One. 2012; 7(2): e30427.Article Pubmed
[66]Jin HO, Lee YH, Park JA, Kim JH, Hong SE, et al. Blockage of Stat3 enhances the sensitivity of NSCLC cells to PI3K/mTOR inhibition. Biochem Biophys Res Commun. 2014; 444(4):502–508.Article Pubmed
[67]Shtivelman E, Hensing T, Simon GR, Dennis PA, Otterson GA, et al. Molecular pathways and therapeutic targets in lung cancer. Oncotarget. 2014; 5(6):1392–1433.Article Pubmed
[68]Kurman RJ. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Annals Oncol. 2013; 24 (Suppl 10):16–21.Article Pubmed
[69]Koshiyama M, Natsumura N, Konishi I. Recent concepts of ovarian carcinogenesis: type I and type II. Biomed Res Int. 2014; 934261.Article Pubmed
[70]Mahajan A, Liu Z, Gellert L, Zou X, Yang G, et al. HMGA2: a biomarker significantly overexpressed in high-grade ovarian serous carcinoma. Mod Pathol. 2010: 23(5):673–681.Article Pubmed
[71]Masciullo V, Baldassarre G, Pentimalli F, Berlingieri MT, Boccia A, et al. HMGA1 protein over-expression is a frequent feature of epithelial ovarian carcinomas. Carcinogenesis. 2003; 24(7):1191–1198.Article Pubmed
[72]Peters DG, Kudla DM, DeLoia JA, Chu TJ, Fairfull L, et al. Comparative gene expression analysis of ovarian carcinoma and normal epithelium by serial analysis of gene expression. Cancer Epidemiol Biomarkers Prev. 2005; 14(7):1717–1723.Article Pubmed
[73]Malek A, Bakhidze E, Noske A, Sers C, Aigner A, et al. HMGA2 gene is a promising target for ovarian cancer silencing therapy. Int J Cancer. 2008; 123(2):348–356.Article Pubmed
[74]Wu J, Liu Z, Shao C, Gong Y, Hermando E, et al. HMGA2 overexpression-induced ovarian surface epithelial transformation is mediated through regulation of EMT genes. Cancer Res. 2011; 71(2):349–359.Article Pubmed
[75]Hetland TE, Holt A, Kærn J, Flørenes VA, Tropé CG, et al. HMGA2 protein expression in ovarian serous carcinoma effusion, primary tumors, and solid metastases. Virchows Arch. 2012; 460(5):505–513.Article Pubmed
[76]Park SM, Shell S, Radjabi AR, Schickel R, Feig C, et al. Let-7 prevents early cancer progression by suppressing expression of the embryonic gene HMGA2. Cell Cycle. 2007; 6(21):2585–2590.Article Pubmed
[77]Montserrat N, Mozos A, Llobe D, Dolcet X, Pons C, et al. Epithelial to mesenchymal transition in early stage endometrioid endometrial carcinoma. Human Pathol. 2012; 43(5):632–643.Article Pubmed
[78]He X, Yang J, Zhang Q, Cui H, Zhang Y. Shortening of the 3'untranslated region: An important mechanism leading to overexpression of HMGA2 in serous ovarian carcinoma. Chin Med J. 2014; 127(3):494–499.Article Pubmed
[79]Liu Z, Liu J, Segura MF, Shao C, Lee P, et al. Mir-182 overexpression in tumorigenesis of high-grade serous ovarian carcinoma. J Pathol. 2012; 228(2):204–215.Article Pubmed
[80]McMillen BD, Aponte MM, Liu Z, Helenowski IB, Scholtens DM, et al. Expression analysis of MIR182 and its associated target genes in advanced ovarian carcinoma. Mod Pathol. 2012; 25(12):1644–1653.Article Pubmed
[81]De Marco C, Rinaldo N, Bruni P, Malzoni C, Zullo F, et al. Multiple genetic alterations within the PI3K pathway are responsible for AKT activation in patients with ovarian carcinoma. PLoS One. 2013; 8(2):e55362.Article Pubmed
[82]Stelniec-Klotz I, Legewie S, Tchernitsa O, Witzel, F, Klinger B, et al. Reverse engineering a hierarchical regulatory network downstream of oncogenic KRAS. Mol Syst Biol. 2012; 8:601.Article Pubmed
[83]Chiappetta G, Tallini G, De Blasio C, Manfioletti G, Martinez-Tello F, et al. Detection of high mobility group HMGI(Y) protein in the diagnosis of thyroid tumours: HMGI(Y) expression represents a potential diagnostic indicator of carcinoma. Cancer Res. 1998; 58(18):4193–4198.Article Pubmed
[84]Czyż W, Balcerczak E, Jakubiak M, Pasieka Z, Kuzdak K, et al. HMGI(Y) gene expression as a potential marker of thyroid follicular carcinoma. Langenbecks Arch Surg. 2004; 389(3):193–197.Article Pubmed
[85]Martinez Hoyos J, Ferraro A, Sacchetti S, Keller S, De Martino I, et al. HAND1 gene expression is negatively regulated by the high mobility group A1 proteins and is drastically reduced in human thyroid carcinomas. Oncogene. 2009; 28(6):876–885.Article Pubmed
[86]Frasca F, Rustighi A, Malaguarnera R, Altamura S, Vigneri P, et al. HMGA1 inhibits the function of p53 family members in thyroid cancer cells. Cancer Res. 2006; 66(6):2980–2989.Article Pubmed
[87]Berlingieri MT, Pierantoni GM, Giancotti V, Santoro M, Fusco A. Thyroid cell transformation requires the expression of the HMGA1 protein. Oncogene. 2002; 21(19):2971–2980.Article Pubmed
[88]Mussnich P, D’Angelo D, Leone V, Croce CM, Fusco A. The high mobility group A proteins contribute to thyroid cell transformation by regulating miR-603 and miR-10b. Mol Oncol. 2013; 7(3):531–542.Article Pubmed
[89]Wei CH, Wei LX, Lay MY, Chen JZ, Mo XJ. Effect of silencing of the high mobility group A2 gene on gastric cancer MKN-45 cells. World J Gastroenterol. 2013; 19(8):1239–1246.Article Pubmed
[90]Shah NR, Chen H. MicroRNAs in pathogenesis of breast cancer: Implications in diagnosis and treatment. World J Clin Oncol. 2014; 5(2):48–60.Article Pubmed
[91]Belge G, Meyer A, Klemke M, Burchardt K, Stern C, et al. Upregulation of HMGA2 in thyroid carcinomas: a novel molecular marker to distinguish between benign and malignant follicular neoplasias. Genes Chromosomes Cancer. 2008; 47(1):56–63.Article Pubmed
[92]Chiappetta G, Ferraro A, Vuttariello E, Monaco M, Galdiero F, et al. HMGA2 mRNA expression correlates with the malignant phenotype in human thyroid neoplasias. Eur J Cancer. 2008; 44(7):1015–1021.Article Pubmed
[93]Jin L, Lloyd RV, Narrar A, Lappinga PJ, Sebo TJ, et al. HMGA2 expression analysis in cytological and paraffin-embedded tissue specimens of thyroid tumors by relative quantitative RT-PCR. Diagn Mol Pathol. 2011; 20(2):71–80.Article Pubmed
[94]Lappinga PJ, Kip NS, Jin L, Jin L, Lloyd RV, et al. HMGA2 gene expression analysis performed on cytologic smears to distinguish benign from malignant thyroid nodules. Cancer Cytopathol. 2010; 118(5):287–297.Article Pubmed
[95]Prasad NB, Kowalski J, Tsai H-L, Talbot K, Somerven H, et al. Three-gene molecular diagnostic model for thyroid cancer. Thyroid. 2012; 22(3):275–284.Article Pubmed
[96]Donato G, Martine Hoyos J, Amorosi A, Maltese L. High mobility group A1 expression correlates with the histological grade of human glial tumors. Oncol Rep. 2004; 11(6):1209–1213.Article Pubmed
[97]Lau KM, Chan QK, Pang JC, Ma FM, Li KK, et al. Overexpression of JMGA1 deregulates tumor growth via cdc25A and alters migration/invasion through cdc25A-independent pathway in medulloblastoma. Acta Neuropathol. 2012. 123(4):553–571.Article Pubmed
[98]Pang B, Fan H, Zhang IY, Liu B, Feng B, et al. HMGA1 expression in human gliomas and its correlation with tumor proliferation, invasion and angiogenesis. J Neurooncol. 2012; 106(3):543–349.Article Pubmed
[99]Shen T, Huang S. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anti-cancer Agents Med Chem. 2012; 12(6):631–639.Article Pubmed
[100]Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit Rev Biochem Mol Biol. 2013; 48(3):222–272.Article Pubmed
[101]Labrie M, St-Pierre Y. Epigenetic regulation of mmp-9 gene expression. Cell. Mol Life Sci. 2013; 70(17):3109–3124.Article Pubmed
[102]Gong J, Zhu S, Zhang Y, Wang J. Interplay of VEGFa and MMP2 regulated invasion of glioblastoma. Tumour Biol. 2014; 35(12):11879–11885.Article Pubmed
[103]Fernández JG, Rodríguez D, Valenzuela M, Calderon C, Urzúa U, et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced β-catenin/Tcf-Lef dependent transcription. Mol Cancer. 2014; 13:209.Article Pubmed
[104]Wang P, Zhen H, Zhang J, Zhang W, Zhang R, et al. Survivin promotes glioma angiogenesis through vascular endothelial growth factor and basic fibroblast growth factor in vitro and in vivo. Mol Carcinog. 2012; 51(7):586–595.Article Pubmed
[105]Wu T, Jia J, Xiong X, He H, Bu L, et al. Increased expression of Lin28B associates with poor prognosis in patients with oral squamous cell carcinoma. PLoS One. 2013; 8(12):e83869.Article Pubmed
[106]Liu B, Pang B, Hou X, Fan H, Liang N, et al. Expression of high-mobility group AT-hook protein 2 and its prognostic significance in malignant glioma. Human Pathol. 2014; 45(8):1752–1758.Article Pubmed
[107]Yang MY, Chen MT, Huang PI, Wang CY, Chang YC, et al Nuclear localization signal-enhanced polyurethane-short branch polyethylenimine-mediated delivery of let-7a inhibited cancer stem-like properties by targeting the 3'-UTR of HMGA2 in anaplastic astrocytoma. Cell Transplant. 2015; 24(8):1431–1450.Article Pubmed
[108]Han X, Fang X, Lou X, Hua D, Ding W, et al. Silencing SOX2 induced mesenchymal-epithelial transition and its expression predicts liver and lymph node metastasis of CRC patients. PLoS One. 2012; 7(8):e41335.Article Pubmed
[109]Wang XR, Luo H, Li HL, Cao L, Wang XF, et al. Overexpressed let-7a inhibits glioma cells malignancy by directly targeting K-ras, independently of PTEN. Neuro-Oncology. 2013; 15(11):1491–1501.Article Pubmed
[110]Mao X-g, Hütt-Cabezas M, Orr BA, Weingart M, Taylor I, et al. LIN28A facilitates the transformation of human neural stem cells and promotes glioblastoma tumorigenesis through a pro-invasive genetic program. Oncotarget. 2013; 4(7):1050–1064.Article Pubmed
[111]Nishino J, Kim S, Zhu Y, Zhu H, Morrison SJ. A network of heterochronic genes including Imp1 regulates temporal changes in stem cells properties. eLife. 2013; 2:e00924.Article Pubmed
[112]Fujii Y, Kishi Y, Gotoh Y. IMP2 regulates differentiation potentials of mouse neocortical neural precursor cells. Genes Cells. 2013; 18(2):79–89.Article Pubmed
[113]Janiszewska M, Suvà ML, Riggi N, Houtkooper RH, Provero, P, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012; 26(17):1926–1944.Article Pubmed
[114]Fedele M, Battista S, Manfioletti G, Croce CM, Giancotti V, et al. Role of the high mobility group A proteins in human lipomas. Carcinogenesis. 2001; 22(10):1583–1591.Article Pubmed
[115]Sgarra R, Rustighi A, Tessari MA, Di Bernardo J, Altamura S, et al. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 2004; 574(1–3):1–8.Article Pubmed
[116]Kishi Y, Fujii Y, Hirabayashi Y, Gotoh Y. HMGA regulates the global chromatin state and neurogenic potential of neocortical precursor cells. Nat Neurosci. 2012; 15(8):1127–1133.Article Pubmed
[117]Ozturk N, Singh I, Mehta A, Braun T, Barreto G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front Cell Dev Biol. 2014; 2:5.Article Pubmed
[118]Chen Z, Cheng Q, Ma Z, Xi H, Peng R, et al. Overexpression of RKIP inhibits cell invasion in glioma cell lines through up-regulation of miR-98. Biomed Res Int. 2013; 2013:695179.Article Pubmed
[119]Chiou GY, Chien CS, Wang ML, Chen MT, Yang YP, et al. Epigenetic regulation of the miR 142-3p/interleukin-6 circuit in glioblastoma. Mol Cell. 2013; 52(5):693–706.Article Pubmed
[120]Carraro G, Shrestha A, Rostkovius J, Contreras A, Chao CM, et al. miR-142-3p balances proliferation and differentiation of mesenchymal cells during lung development. Development. 2014; 141(6):1272–1281.Article Pubmed
[121]Luwor RB, Stylli SS, Kaye AH, The role of Stat3 in glioblastoma multiforme. J Clin Neurosci. 2013; 20(7):907–911.Article Pubmed
[122]Stechishin OD, Ha L, Ruan Y, Blough MD, Nguyen SA, et al. On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro-Oncol. 2013; 15(2):198–207.Article Pubmed
[123]Ashizawa T, Miyata H, Iizuka A, Komiyama M, Oshita C, et al. Effect of STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int J Oncol. 2013; 43(1):219–227.Article Pubmed
[124]Møller HG, Rasmussen AP, Andersen HH, Johnsen KB, Henriksen M, et al. A systematic review of microRNA in glioblastoma multiforme: micro-modulators in the mesenchymal mode of migration and invasion. Mol Neurobiol. 2013; 47(1):131–144.Article Pubmed
[125]Chu PM, Ma HI, Chen LH, Chen MT, Huang PI, et al. Deregulated microRNAs identified in isolated glioblastoma stem cells: an overview. Cell Transplant. 2013; 22(4):741–753.Article Pubmed
[126]Li Y, Guessous F, Zhang Y, DiPierro C, Kefas B, et al. microRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009; 69(19):7569–7576.Article Pubmed
[127]Rokavec M, Li H, Jiang L, Hermeking H. The p53/miR34 axis in development and disease. J Mol Cell Biol. 2014; 6(3):214–230.Article Pubmed
[128]Cao W, Fan R, Wang L, Cheng S, Li H, et al. Expression and regulatory function of miRNA-34a in targeting survivin in gastric cancer cells. Tumour Biol. 2013; 34(2):963–971.Article Pubmed
[129]Cao W, Yang W, Fan R, Li H, Jiang J, et al. miR-34a regulates cisplatin-induce gastric cancer cell death by modulating PI3K/AKT/surviving pathway. Tumour Biol. 2014; 35(2):1287–1295.Article Pubmed
[130]Viswanathan S, Powers JT, Einhorn W, Hoshida Y, Ng TL, et al. Lin 28 promotes transformation and is associated with human advanced malignancies. Nat Genet. 2009; 41(7):843–848.Article Pubmed
[131]Gaytan F, Sangiao-Alvarellos S, Manfredi-Lozano M, Garcia-Galiano D, Ruiz-Pino F. et al. Distinct expression patterns predict differential roles of the miRNA-binding proteins, Lin28 and Lin28b, in the mouse testis: studies during postnatal development and in a model of hypogonadotropic hypogonadism. Endocrinology. 2013; 154(3):1321–1336.Article Pubmed
[132]Zhou J, Ng SB, Chng WJ. LIN28/LIN28B: An emerging oncogenic driver in cancer stem cells. Int J Biochem Cell Biol. 2013; 45(5):973–978.Article Pubmed
[133]Zhang K, Gao H, Xu X, Wang J, Zhou W, et al. Frequent overexpression of HMGA2 in human atypical teratoid/rhabdoid tumor and it correlation with let-7a3/let-7b miRNA. Clin Cancer Res. 2014; 20(5):1179–1189.Article Pubmed
[134]Zhu H, Shah S, Shyh-Chang N, Shinoda G, Einhorn WS, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat Genet. 2010; 42(7):626–630.Article Pubmed
[135]Piskounova E, Polytarchou C, Thomton JE, LaPierre RJ, Pothoulakis C, et al. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell. 2011; 147(5):1066–1079.Article Pubmed
[136]Sgarra R, Zammitti S, Lo Sardo A, Maurizio E, Arnoldo L, et al. HMGA molecular network: From transcriptional regulation to chromatin remodelling. Biochimi Biophys Acta. 2010; 1799(1–2):37–47.Article Pubmed
[137]Shlapobersky M, Sanders R, Clark C, Spector DH. Repression of HMGA2 gene expression by human cytomegalovirus involves the IE2 86-kilodalton protein and is necessary for efficient viral replication and inhibition of Cyclyn A transcription. J Virol. 2006; 80(20):9951–9961.Article Pubmed
[138]Tessari MA, Gostissa M, Altamura S, Sgarra R, Rustighi A, et al. Transcriptional activation of the cyclin A gene by the architectural transcription factor HMGA2. Mol Cell Biol. 2003; 23(24):9104–9116.Article Pubmed
[139]Chiappetta G, Manfioletti G, Pentimalli F, Abe N, Di Bonito M, et al. High mobility group HMGI(Y) protein expression in human colorectal hyperplastic and neoplastic diseases. Int J Cancer. 2001; 91(2):147–151.Article Pubmed
[140]Berlingieri MT, Manfioletti G, Santoro M, Bandiera A, Visconti R, et al. Inhibition of HMGI-C protein synthesis suppresses retroviral induced neoplastic transformation of rat thyroid cells. Mol Cell Biol. 1995; 15(3):1545–1553.Article Pubmed
[141]Zhou X, Benson KF, Ashar HR, Chada K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature. 1995; 376(6543):771–774.Article Pubmed
[142]Federico A, Forzati F, Esposito F, Arra C, Palma G, et al. Hmga1/Hmga2 double knock-out mice display a “superpygmy” phenotype. Biol Open. 2014; 3(5):372–378.Article Pubmed
[143]Yang N, Hui L, Wang Y, Yang H, Jiang X. Overexpression of SOX2 promotes migration, invasion, and epithelial-mesenchymal transition through the Wnt/β-catenin pathway in laryngeal cancer Hep-2 cells. Tumour Biol. 2014; 35(8):7965–7973.Article Pubmed
[144]Chien CS, Wang ML, Chu PY, Chang YL, Liu WH, et al. Lin28B/Let-7 regulates expression of Oct4 and Sox2 and reprograms oral squamous cell carcinoma cells to a stem-like state. Cancer Res. 2015; 75(12):2553–2565.Article Pubmed
[145]Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci USA. 2009; 106(9):3384–3389.Article Pubmed
[146]Watanabe S, Ueda Y, Akaboshi S, Hino Y, Sekita Y, et al. HMGA2 maintains oncogenic Ras-induced epithelial mesenchymal transition in human pancreatic cancer cells. Am J Pathol. 2009; 174(3):854–868.Article Pubmed
[147]Kolenda T, Przybyta W, Teresiak A, Mackiewcz A, Lamperska KM. The mystery of let-7d: A small RNA with great power. Contemp Oncol. 2014; 18(5):293–301.Article Pubmed
[148]Wagner S, Ngezahayo A, Murua Escobar H, Nolte I. Role of miRNA let-7 and its major targets in prostate cancer. Biomed Res Int. 2014; 2014:376326.Article Pubmed
[149]Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzhnrubl D, et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013; 5(8):1196–1212.Article Pubmed
[150]Romero-Perez L, Lopez-Garcia MA, Diaz-Martin J, Biscuola M, Castilla MA, et al. ZEB1 overexpression associated with E-cadherin and microRNA-200 downregulation is characteristic of undifferentiated endometrial carcinoma. Mod Pathol. 2013; 26(11):1514–1524.Article Pubmed
[151]Gheldof A, Hulpian P, vanRoy F, De Craene B, Berx G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol Life Sci. 2012; 69(15):2527–2541.Article Pubmed
[152]Tan EJ, Kahata K, Idas O, Thuault S, Heldin CH, et al. The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial-to-mesenchymal transition. Nucleic Acids Res. 2015; 43(1):162–178.Article Pubmed
[153]Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, et al. Functional cooperation between Snail1 and Twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011; 286(14):12024–12032.Article Pubmed
[154]Song X, Liu W, Xi S, Wang M, Cao G, et al. All-transretinoic acid ameliorates bleomycin-induced lung fibrosis by downregulating the TGF-β1/Smad3 signaling pathway in rats. Lab Invest. 2013; 93(11):1219–1231.Article Pubmed
[155]Zhang P, Huang C, Fu C, Tian Y, Hu Y, et al. Cordycepin (3’-deoxyadenosine) suppressed HMGA2, Twist1 and ZEB1-dependent melanoma invasion and metastasis by targeting miR-33b. Oncotarget. 2015; 6(12):9834–9853.Article Pubmed
[156]Franco HL, Casanovas J, Rodriguez-Medina JR, Cadilla CL. Redundant or separate entities?--roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011; 39(4):1177–1186.Article Pubmed
 Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved