



- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Cancer Research & Therapy
Volume 1, Issue 4, June 2013, Pages 138-142
Case reportOpen Access
Therapy-related acute myeloid leukemia with chromosomal abnormalities involving t(9;22)(q34;q11) and t(3;21)(q26;q22) during chemotherapy for follicular lymphoma
-
Ogasawara T1,3,*
 ,
Aiba M2 and
Kawauchi K1,4
,
Aiba M2 and
Kawauchi K1,4
*Corresponding author: Ogasawara T, Department of Medicine, Tokyo Women’s Medical University, Medical Center East, 2-1-10 Nishiogu Arakawa-ku, Tokyo, 116-8567, Japan, Tel.: 81-3-3964-1141; Fax: 81-3-3964-1982. E-mail: toshie_ogasawara@tmghig.jp
Received 17 March 2013 Revised 27 April 2013 Accepted 5 May 2013; Published 15 May 2013
DOI: http://dx.doi.org/10.14312/2052-4994.2013-21
Copyright: ©2013 Ogasawara T, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
This report describes a case of a patient who developed therapy-related acute myeloid leukemia five years after initiating chemotherapy for follicular lymphoma. The patient had been treated with multiple chemotherapeutic regimens, including anthracycline and etoposide (VP-16), as well as with radiation therapy for refractory follicular lymphoma over the preceding five years. The patient subsequently developed myelodysplastic syndrome (MDS) with karyotypic abnormalities of monosomy 7 and del (q11; q13.3) followed by acute myeloid leukemia (AML) with an additional balanced translocation of t(9;22)(q34;q11) and t(3;21)(q26;q22). Reverse transcription-polymerase chain reaction amplification of the patient’s RNA showed a fusion transcript of minor BCR-ABL but not EVI1-RUNX1 (AML1) genes. Imatinib therapy resulted in regression of AML, but the patient soon became refractory to chemotherapy and died. Therapy-related acute leukemia develops mostly as non-lymphoid leukemia with unbalanced aberrations of monosomy 7 and 5 or balanced aberrations involving 11q23 and 21q22, but Philadelphia chromosome is uncommon. In addition, simultaneous occurrence of both t(9;22)(q34;q11) and t(3;21)(q26;q22) balanced aberrations in t-MDS/t-AML is a very rare event. The balanced translocations detected in this case suggest another mechanism by which t-AML can develop after chemotherapy and radiation therapy for follicular lymphoma.
Keywords: therapy-related myelodysplastic syndrome (t-MDS); therapy-related acute myeloid leukemia (t-AML); t(9;22); t(3;21); Philadelphia chromosome; follicular lymphoma; imatinib; chemotherapy
IntroductionTop
Chemotherapy or radiation therapy for hematological malignancies and various solid tumors can cause myelodysplastic syndrome or acute non-lymphoid leukemia, defined as therapy-related myelodysplastic syndrome/therapy-related acute myeloid leukemia (t-MDS/t-AML). Acute myeloid leukemia following chemotherapy was initially described by Sypkens et al. in 1970 [1] and, in the 1980s, alkylating agents was recognized as causative agent of t-MDS/AML [2] . In the 2008 edition of the World Health Organization (WHO) classification [3] , t-MDS/AML and therapy-related myeloproliferative neoplasms (t-MPN) were defined as therapy-related myeloid neoplasms (t-MN), which occur as late complications of cytotoxic chemotherapy and/or ionizing radiation therapy used for the treatment of various neoplasms and non-neoplasms. These entities were included within the group of acute myeloid leukemia (AML). t-MN can be classified into two different types according to the causative cytotoxic agent. The first type, which usually presents as t-MDS, is associated with alkylating agents with unbalanced abnormalities of chromosomes 5 and 7. The second type, which presents as t-AML without preceding myelodysplasia, is associated with DNA topoisomerase II inhibitors, such as etoposide (VP-16) and anthracycline, with typically balanced translocations involving rearrangement of 11q23 or 21q22. t-AML accompanied by t(9;22)(q34;q11) called Philadelphia (Ph) chromosome has been documented [4] but is a rare event.
The present case describes a case of t-MDS with monosomy 7 followed by t-AML accompanied by chromosomal translocations of t(9;22)(q34;q11) and t(3;21)(q26;q22) after chemotherapy and radiation therapy for follicular lymphoma, suggesting a causative role of this therapy in the transition from t-MDS to t-AML.
Case reportTop
A 60-year-old man presented to our hospital with abdominal distention in November 2000. His past medical history, social history, and family history were unremarkable. Physical examination showed marked hepatosplenomegaly (liver edge palpable at five fingers below the right costal margin, and spleen palpable at 10 fingers below the left costal margin) and superficial lymph nodes that were palpable at the cervical, axillar, and inguinal regions. His body temperature was >38 ℃. Computed tomography scan revealed systemic lymphadenopathies, hepatomegaly, and massive splenomegaly. Laboratory studies revealed a white blood cell (WBC) count of 7.8 x 109/L with a normal differential, hemoglobin (Hb) of 10.0 g/dL, platelet count of 16 x 109/L, lactate dehydrogenase of 253 IU/L (normal range, 142-246), and soluble interleukin-2 receptor level of 13,900 U/mL (normal range, 188-570). Excisional biopsy of the left inguinal lymph node revealed predominantly proliferated centrocytes with a follicular pattern, and cells were positive for CD20, CD10, and Bcl-2 and showed t(14;18)(q32;q21) with fusion transcript between immunoglobulin heavy chain and Bcl-2 (Figure 1). Bone marrow examination showed lymphoma cells with normal karyotype. Follicular lymphoma (grade 1), stage IVB was diagnosed with an intermediate risk according to Follicular Lymphoma International Prognostic Index (FLIPI). The patient underwent eight cycles of chemotherapy, consisting of cyclophosphamide (CPA), doxorubicin hydrochloride (ADR), vincristine (VCR), prednisolone (PSL) and VP-16, and then achieved a partial remission. He subsequently underwent radiation therapy (29 Gy) for his massive splenomegaly. In February 2002, he was treated with oral VP-16 for enlarged cervical lymph nodes. In August 2003, he underwent chemotherapy with rituximab, CPA, ADR, VCR, and PSL for relapsed follicular lymphoma, which resulted in regression of the lymphoma. However, left cervical lymphadenopathy recurred in March 2004, and he received radiation therapy. In January 2005, he underwent four cycles of chemotherapy with dexamethasone, VP-16, ifosfamide (IFM), and carboplatin (CBDCA) due to systemic lymphadenopathies with minimal response. In September 2005, he developed pancytopenia, and his bone marrow showed trilineage dysplasia accompanied by chromosomal aberrations of monosomy 7 and del(20)(q11;q13.3), suggesting the emergence of t-MDS. Because his lymphoma progressed in an aggressive fashion, he was treated with mitoxantrone (MIT), VP-16, and PSL. In February 2006, his peripheral blood showed a WBC count of 156,000/µL with 45% blasts, and bone marrow aspirate showed diffuse proliferation of blasts (45% in all nucleated cells), which were positive for myeloperoxidase (Figure 2A). Cytogenetic analysis of bone marrow revealed additional karyotypic aberrations of t(9;22)(q34;q11) and t(3;21)(q26;q22) in 20 of 20 cells analyzed (Figure 2B). Flow cytometric analysis of bone marrow revealed expression of CD7 (81.7%), CD20 (11.3%), CD13 (77.8%), CD33 (56.9%), CD34 (88%), and HLA-DR (86.3%). Peripheral blood blasts represented fusion transcripts of minor BCR-ABL (472 bp) but not AML1-EVI1 (587 bp) according to polymerase chain reaction (PCR) analysis (Figure 2C). A diagnosis of t-AML was made. Surprisingly, administration of imatinib resulted in marked reduction of blasts, while low-dose cytarabin therapy was ineffective. After recovering from severe cytopenia, imatinib was resumed, but the leukemia failed to improve. Chemotherapy containing MIT, Ara-C, and VCR was ineffective, and he died in May 2006. Cumulative doses of CPA, ADR, VCR, MIT, VP-16, IFM, CBDCA, and Ara-C were 18,450 mg, 884 mg, 30 mg, 128 mg, 11,915 mg, 24,000 mg, 1,740 mg, and 2,505 mg, respectively.
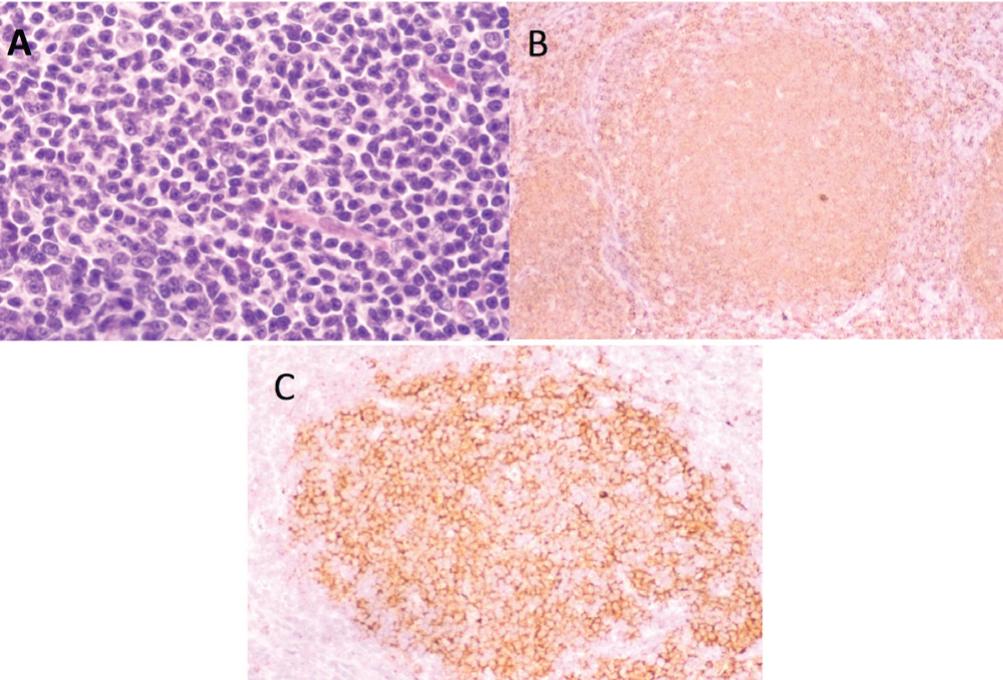
Figure 1 Morphology of lymph node biopsy at initial presentation. (A) Histopathology examination of the lymph node shows follicular lymphoma (hematoxylin and eosin stain); (B) Lymphoma cells are positive for CD20; (C) Lymphoma cells are positive for BCL-2.
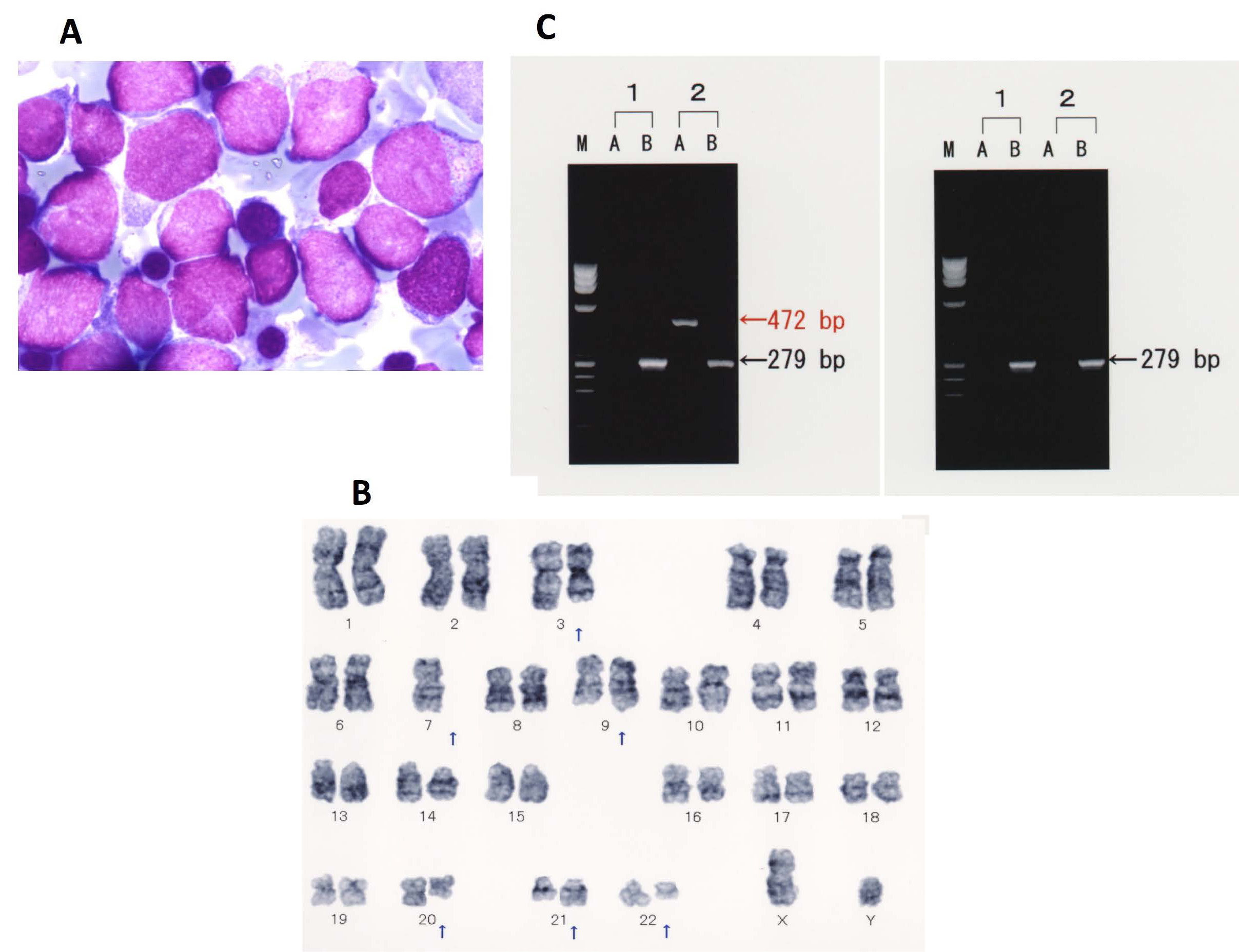
Figure 2 Morphological, cytogenetic, and molecular analyses of bone marrow aspirate at the development of t-AML. (A) Leukemia cells in bone marrow (Wright-Giemsa stain); (B) Chromosomal aberrations in bone marrow showed t(9;22)(q32;q11), t(3;21)(q26;q22), and monosomy 7; (C) RT-PCR analysis revealed a minor BCR-ABL fusion gene (left panel): 1) Negative control; 2) Patient, M. size marker, A. minor BCR-ABL (472 bp), B. β-actin (279 bp); RT-PCR analysis revealed no AML1-EVI1 fusion gene (right panel): 1) Negative control; 2) Patient, M. size marker, A. AML1-EVI1 (587 bp), B. β-actin (279 bp).
DiscussionTop
t-MN associated with alkylating agents and radiation therapy usually develops after a long latency period of more than five years following chemotherapy. In that case, t-AML transforms from t-MDS with chromosomal aberration of monosomy 5 or 7. The present case developed MDS (t-MDS) with monosomy 7, five years after beginning chemotherapy for follicular lymphoma; this t-MDS was compatible with the characteristics of t-MN associated with alkylating agents. Afterwards, the patient progressed to AML (t-AML) with the presence of Ph chromosome of t(9;22)(q34;q11) and t(3;21)(q26;q22), in addition to monosomy 7. These data suggest that Ph chromosome and t(3;21)(q26;q22) play a role in the transformation of t-MDS to t-AML. Such balanced chromosomal aberrations could be associated with the use of topoisomerase II inhibitors, such as VP-16 or anthracyclines. Topoisomerase II inhibitors can cause genomic instability and result in double-strand breakage (DSB) by forming a complex with topoisomerase and DNA (DNA breakage-first theory) [5] , which is repaired mainly by non-homologous end joining (NHEJ) [6] . When damaged cells carrying DSBs pass through the check point of the G2/M stage in the cell cycle, balanced translocations can be induced. Pederson-Bjergaard et al. [7] reported that VP-16 alone or in combination with cisplatin increased the risk of secondary leukemia. Further, patients who received VP-16 at a cumulative dose of greater than 2,000 mg/m2 had an increased risk of developing AML. In the present case, the cumulative total dose of VP-16 was approximately 7,000 mg/m2, suggesting that VP-16 is a causative agent for AML following t-MDS.
Ph chromosome is a rare chromosome abnormality in t-MN. Andersen et al. [8] described the occurrence of Ph chromosome in secondary MDS and AML in Mitelman’s Catalog and a significant association between Ph chromosome and topoisomerase II inhibitors. In their study, patients mostly had non-hematological malignancies; in fact, only two cases had hematological malignancies, including one case of non-Hodgkin lymphoma. Block et al. [9] analyzed 511 cases of t-MDS and t-AML that were registered into the international workshop (US, Europe, and Japan in 2002), including 10 cases with Ph chromosome, which comprised five cases of t-AML and five cases of therapy-related acute lymphoblastic leukemia (t-ALL). However, all cases with Ph chromosome occurred without preceding MDS, and only one case had both monosomy 5 and 7. In contrast, the present case presented with an MDS phase prior to AML and was associated with t(9;22)(q34;q11), which generated a minor BCR-ABL fusion transcript, as confirmed by PCR. Minor BCR-ABL transcripts have been previously described in only one case with Ph chromosome among malignant lymphoma [10] . In another case, Nakane et al. described a patient who developed t-AML from multiple myeloma, showing a BCR-ABL fusion gene without Ph chromosome by G-banding [11] . Case reports [10, 12, 13, 14] of therapy-related acute leukemia with t(9;22)(q34;q11) in patients with malignant lymphoma as the primary disease are summarized in Table 1. Among these, only one case [10] had follicular lymphoma as the primary tumor, while Hodgkin lymphoma was the most common type of original lymphoma. Cytogenetic analysis has revealed an abnormality involving monosomy 7, which is usually found in MDS, in only one case aside from the present case, which reflects the direct progression from primary disease to t-AML without an MDS phase.
| Case reports | F/M | Primary tumor | -7 | Chromosomes | Therapy | t-AL | Latent (months) | |||
| t(9;22) | t(3;21) | Topo II | Alk | RT | ||||||
| 10 | M | NHL (nodular) | + | +# | - | + | + | - | AML | 123 |
| 12 | M | HL | - | + | - | + | + | + | ALL | 19 |
| 13 | M | HL | - | + | - | + | + | + | AML (M3) | 16 |
| 14 | F | NHL (diffuse) | - | + | - | + | + | - | AML (M3) | 10 |
| Present case | M | NHL (follicular) | + | + | + | + | + | + | AML | 3 |
Abbreviations: #: Variant Ph, HL: Hodgkin lymphoma, NHL: non-HL, Topo II: topoisomerase II inhibitor, including VP16 and anthracycline, Alk: alkylating agent, RT: radiation therapy, t-AL: therapy-related acute leukemia, M: male, F: female
In addition to t(9;22)(q34;q11), we observed the emergence of t(3;21)(q26;q22), which is also found in t-MDS and t-AML. The incidence of 21q22 balanced chromosomal abnormalities is estimated as 15.5% in t-MDS/t-AML, according to the international workshop in 2002 [15] . The balanced t(3;21)(q26;q22) aberration was found in 20% of cases associated with 21q22. The t(3;21)(q26;q22) generates a fusion gene between AML1/RUNX1 and either RPL22L1, MDS1, or EVI1 [16] ; however, an analysis of such a fusion gene has been performed in only a few t-MDS/t-AML cases [15] . While we found the balanced t(3;21)(q26;q22), we could not detect the AML1/RUNX1-EVI1 fusion transcript by PCR analysis. Because we did not examine the MDS1 or PRL22L1 fusion transcripts, the involvement of these genes in this translocation cannot be excluded. The t(3;21)(q26;q22) aberration including the AML1/RUNX1 rearrangement also appears to be associated with topoisomerase inhibitors in t-MDS/t-AML (17), indicating that t(3;21)(q26;q22) in concert with t(9;22)(q34;q11) (which could be induced by topoisomerase II inhibitors) might have induced t-AML in the present case. BCR-ABL chimeric protein generated by the t(9;22)(q34;q11) reciprocal translocation has constitutive tyrosine kinase activity and activates downstream signal transduction pathways, including Ras/mitogen-activated protein kinase, phosphatidylinositol 3-kinase/Akt/mechanical target of rapamycin, and Janus kinase/signal transducer and activator of transcription, leading to promotion of leukemic cell proliferation and survival [18] . Imatinib mesylate, a small molecule tyrosine kinase inhibitor that directly targets BCR-ABL, induces hematological and cytogenetic remissions in most chronic myeloid leukemia (CML) patients in the chronic phase. On the other hand, the RUNX1 protein encoded by RUNX1 gene on chromosome 21q22 and the EVI1 protein encoded by EVI1 gene on chromosome 3q26.5, both of which are nuclear transcription factors, play critical roles in hematopoietic stem cell maturation and proliferation, respectively [19] . The RUNX1-EVI1 chimeric protein generated by the t(3;21)(q26;q22) translocation, which represses the function of wild-type RUNX1 protein, interferes with myeloid cell maturation and transforms bone marrow progenitor cells. Furthermore, aberrant expression of RUNX1 or RUNX1-EVI1 fusion gene induces acute transformation of chronic phase as well as imatinib-resistance in CML patients [20] . The fact that imatinib therapy resulted in only transient resolution in this t-AML suggests an important role of t(3;21)(q26;q22) aberration in leukemogenesis.
The coexistence of t(9;22)(q34;q11) and t(3;21)(q26;q22) in t-MN has not previously been reported. The emergence of such translocations in t-AML is associated with high aggressivity and very poor prognosis [9, 15]. Thus, it is important to note the presence of these translocations in the early phase after chemotherapy and to consider more effective treatments, such as stem cell transplantation for eligible t-MN cases.
Conflict of interest
The authors wish to express that they have no conflict of interest.
ReferencesTop
[1] Smit CG, Meyler L (1970) Acute myeloid leukaemia after treatment with cytostatic agents. Lancet 26:671–672. Article Pubmed
[2] Le Beau MM, Albain KS, Larson RA, Vardiman JW, Davis EM, et al. (1986) Clinical and cytogenetic correlations in 63 patients with therapy-related myelodysplastic syndromes and acute nonlymphocytic leukemia: further evidence for characteristic abnormalities of chromosomes no. 5 and 7. J Clin Oncol 4:325–345. Article Pubmed
[3] Vardiman JW, Arber DA, Brunning RD, Larson RA, Matutes E, et al. (2008) Therapy-related myeloid neoplasms. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: International Agency for Research on Cancer pp 129–129.
[4] Pedersen-Bjergaard J, Brøndum-Nielsen K, Karle H, Johansson B (1997) Chemotherapy-related - late occurring - Philadelphia chromosome in AML, ALL and CML. Similar events related to treatment with DNA topoisomerase II inhibitors Leukemia 11:1571–1574. Article Pubmed
[5] Aten JA, Stap J, Krawczyk PM, van Oven CH, Hoebe RA, et al. (2004) Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 303:92–95. Article Pubmed
[6] Povirk LF (2006) Biochemical mechanisms of chromosomal translocations resulting from DNA double-strand breaks. DNA Repair 5:1199–1212. Article Pubmed
[7] Pedersen-Bjergaard J, Daugaard G, Hansen SW, Philip P, Larsen SO, et al. (1991) Increased risk of myelodysplasia and leukaemia after etoposide, cisplatin, and bleomycin for germ-cell tumours. Lancet 338:359–363. Article Pubmed
[8] Andersen MK, Johansson B, Larsen SO, Pedersen-Bjergaard J (1998) Chromosomal abnormalities in secondary MDS and AML. Relationship to drugs and radiation with specific emphasis on the balanced rearrangements. Haematologica 83:483–488. Article Pubmed
[9] Block AW, Carroll AJ, Hagemeijer A, Michaux L, van Lom K, et al. (2002) Rare recurring balanced chromosome abnormalities in therapy-related myelodysplastic syndromes and acute leukemia: report from an international workshop. Genes Chromosomes Cancer 33:401–412. Article Pubmed
[10] Alimena G, Cedrone M, Nanni M, De Cuia MR, Lo Coco F, et al. (1995) Acute leukemia presenting a variant Ph chromosome with p190 expression, dup 3q and -7, developed after malignant lymphoma treated with alkylating agents and topoisomerase II inhibitors. Leukemia 9:1483–1486. Pubmed
[11] Nakase K, Yamamoto Y, Morita K, Yamaguchi T, Nishii K, et al. (2006) Haunting appearance of bcr/abl fusion gene products in a patient with therapy related leukaemia. Leuk Res 30:106–108. Article Pubmed
[12] Yavorkovsky LL, Yavorkovsky LI, Bondare DK (1986) Ph1-positive acute leukemia in a patient with Hodgkin's disease treated by irradiation and chemotherapy. Neoplasma 33:355–359. Pubmed
[13] Dallorso S, Sessarego M, Garré ML, Haupt R, Pasino M, et al. (1990) Secondary acute promyelocytic leukemia with t(8;21) and t(9;22) at onset and loss of the Philadelphia chromosome at relapse. Cancer Cancer Genet Cytogenet 47:41–46. Article Pubmed
[14] Mochiduki Y, Muramoto S (2005) Therapy-related acute promyelocytic leukemia with a t(9;22)(q34;q11) and t(15;17)(q22;q11 to approximately 12) subclone. Rinsho Ketsueki 46:1218–1222. Article Pubmed
[15] Slovak ML, Bedell V, Popplewell L, Arber DA, Schoch C, et al. (2002) 21q22 balanced chromosome aberrations in therapy-related hematopoietic disorders: report from an international workshop. Genes Chromosomes Cancer 33:379–394. Article Pubmed
[16] Park TS, Choi JR, Yoon SH, Song J, Kim J, et al. (2008) Acute promyelocytic leukemia relapsing as secondary acute myelogenous leukemia with translocation t(3;21)(q26;q22) and RUNX1-MDS1-EVI1 fusion transcript. Cancer Genet Cytogenet 187:61–73. Article Pubmed
[17] Bacher U, Schnittger S, Kern W, Trenn G, Weisser M, et al. (2006) Acute myeloid leukemia (AML) with t(8;21)(q22;q22) relapsing as AML with t(3;21)(q26;q22). Cancer Genet Cytogenet 168:172–174. Article Pubmed
[18] Cilloni D, Saglio G (2011) Molecular pathways: BCR-ABL. Clin Cancer Res 18:930–937. Article Pubmed
[19] Maki K, Yamagata T, Mitani K (2008) Role of the RUNX1-EVI1 fusion gene in leukemogenesis. Cancer Sci 99:1878–1883. Article Pubmed
[20] Corm S, Biggio V, Roche-Lestienne C, Laï JL, Yakoub-Agha I, et al. (2005) Coexistence of AML1/RUNX1 and BCR-ABL point mutations in an imatinib-resistant form of CML. Leukemia 19:1991–1992. Article Pubmed
 Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved