



- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Cancer Research & Therapy
Volume 6, Issue 2, February 2018, Pages 6–17
Original researchOpen Access
Combined treatment of gemcitabine with indole-3-carbinol or metformin on drug efficacy in pancreatic cancer cell lines: The role of human equilibrative nucleoside transporters
-
Stancy Joseph1,* Beverly Word1 and
Beverly Lyn-Cook1
 ,
,
*Corresponding author: Beverly D. Lyn-Cook, Division of Biochemical Toxicology, HFT-100, FDA/National Center for Toxicological Research, Jefferson, Arkansas 72079, USA. E-mail: Beverly.lyn-cook@fda.hhs.gov
Received 14 November 2017 Revised 4 January 2018 Accepted 15 January 2018 Published 23 January 2018
DOI: http://dx.doi.org/10.14312/2052-4994.2018-2
Copyright: © 2018 Joseph S, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
Pancreatic cancer is one of the most lethal carcinomas in the United States. In accord with the American Cancer Society pancreatic cancer is anticipated to move from the third to the second leading cause of deaths in the United States by 2020. Although the standard treatment for advanced pancreatic cancer is gemcitabine (GEM), the response rate is less than 20%. Chemoresistance is a hallmark of this cancer, and modulation of drug transporters expression has been shown to increase cancer drug efficacy. Studies have shown that human equilibrative nucleoside transporters (hENTs) expression patterns may predict GEM treatment efficacy. This study investigated whether or not GEM in combination with metformin (MET) or indole-3-carbinol (I3C) increases cytotoxicity and modulates hENT1 and hENT4. Pancreatic cancer cells from males and females were treated for 24 or 72h with GEM and/or MET or I3C. Cell viability, drug interactions, and protein and mRNA expression levels of hENTs were assessed. Treatment with GEM and/or MET or I3C showed cell line specific reductions in pancreatic cancer cell proliferation, and modulation of hENT1 and hENT4 expression. Response to GEM and MET/I3C may be dependent upon the genetic profile of the tumor and the level of expression of a specific transporter. The sensitivity of GEM could depend on the method of treatment, whether cells were pre-treated with MET or I3C, which studies, including our own, have showed that pre-treatment with I3C increased upregulation of hENT1 expression in pancreatic cancer cell lines.
Keywords: pancreatic cancer; gemcitabine; indole-3-carbinol; metformin; human equilibrative nucleoside transporters
IntroductionTop
Pancreatic cancer is the eleventh most common cancer in the US but represents the third leading cause of cancer deaths in men and women [1]. The prognosis is extremely poor, with a 5-years relative survival rate of only 7%, due to the fact that pancreatic cancer is usually asymptomatic in the early stages and most cases are diagnosed at late stages of the disease [1]. Despite significant improvements in understanding molecular and epigenetic changes of pancreatic cancer, the prognosis and management remain unchanged. Treatment and advances in early detection of pancreatic cancer, therefore, remain of critical importance. Effective treatments or novel therapeutic approaches are currently being investigated in clinical trials [2]. Although gemcitabine (GEM) is the gold standard for advance stages of this disease, improvement is needed to enhance further its efficacy for patient survival.
GEM, a deoxycytidine nucleoside analog, is used in advanced pancreatic cancer patients with locally advanced or metastatic cancer [3]. Pancreatic cancer is a highly resistant cancer, due mainly to up regulation of multidrug resistance genes [4, 5]. In addition, patients with various polymorphisms in these genes can have enhanced or decreased efficacy of drugs for treatment [4, 5]. Patients treated with GEM can also develop resistance to this drug. Therefore, it is important to identify other drug agents that, when combined with GEM, will increase the efficacy of GEM.
Previously published data from our laboratory demonstrated enhanced efficacy of GEM with the dietary agent indole-3-carbinol (I3C) in pancreatic cancer [6] and further showed that polymorphisms in ABCB1 in pancreatic cancer cell lines affected GEM efficacy [7]. I3C is a common phytochemical found in cruciferous vegetables. Studies have shown that dietary phytochemicals, such as I3C, may be a promising nontoxic chemopreventive agent with multiple anti-tumor activities, including apoptotic, anti-proliferative, and anti-angiogenic activities [8, 9]. Several clinical trials have investigated the use of I3C for cancer treatment and prevention. In these clinical trials, I3C has been reported to be effective in both the cervix [10] and larynx [11] against precancerous lesions caused by human papillomavirus. In more recent clinical trials, the use of I3C has been investigated in the elimination of cancer-influencing exogenous estrogens in patients with prostate, breast and uterine cancers [8, 12 - 14].
Studies have shown that the expression of several genes may be predictors of GEM efficacy in pancreatic cancer patients [15, 16]. One of the predictor genes for GEM is human equilibrative nucleoside transporter-1 (hENT1), a transporter responsible for the uptake of GEM in human cells [6, 16]. hENT4, another member of the hENT family, has been identified as one of several drug transporters mediating the renal and intestinal uptake of metformin (MET) [17, 18]. MET is the primary drug used for the treatment of type 2 diabetes mellitus, and has become a drug of interest for the treatment of pancreatic cancer [19-23]. Several studies have suggested that patients taking MET for the treatment of diabetes also have a decreased incidence of pancreatic cancer [24, 25]. MET has also been implicated in its ability to increase sensitivity of pancreatic cancer cells to GEM [24]. Other studies have concluded, however, that there is no association between MET use and cancer risk or prognosis [26-27].
This study was performed to evaluate the duo-combined drug effects of GEM and/or MET or I3C in pancreatic cancer cells in relation to chemoresistance. Several pancreatic cancer cell lines were examined for cell viability and drug interactions when treated with GEM, MET, and I3C in mono or combination therapy for 24 or 72 h. We further investigated whether GEM in combination treatment with MET or I3C could modulate hENT1 and hENT4 expression.
Materials and methodsTop
Material
I3C, MET, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), sodium dodecyl sulfate (SDS), hydrochloric acid (HCl), dimethyl sulfoxide (DMSO), bovine serum albumin (BSA), and complete mini protease inhibitor cocktail were purchased from Sigma (St. Louis, MO). GEM was a gift from Eli Lilly (Indianapolis, IN). Fetal bovine serum (FBS), Roswell Park Memorial Institute (RPMI) 1640, Dulbecco’s Minimal Eagle’s Medium (DMEM), phosphate saline buffer (PBS), glutamine, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and sodium pyruvate were purchased from Gibco (Grand Island, NY).
Cell culture
Human pancreatic cancer cell lines from males MIA PaCa2 and PANC1 and females ASPC1, PANC1, and Su.86.86 were obtained from the American Type Culture Collection (ATCC; Manassas, VA). Cell lines were cultured in DMEM (PANC1 and MIA PaCa2) or RPMI 1640 (SU.86.86 and ASPC1) with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin (Invitrogen, Carlsbad, CA) at 37°C in a humidified atmosphere containing 5% CO2. Glutamine, HEPES, and sodium pyruvate supplements were added to maintain proper cell growth for SU.86.86 cells.
MTT assay for cell proliferation
Using the MTT assay, a colormetric assay for measuring cell metabolic activity, cells were seeded in 96-well plates (1 x 104 cells per well), and were treated with therapeutic agents for 24 or 72 h at 37°C in a humidified atmosphere containing 5% CO2. At the end of the treatment period, MTT solution (5 mg/mL) was added to each well (10 µL) and incubated for 4 h at 37°C. Subsequently, a SDS/HCl solution (100 µL) was added. Optical density was determined using a BioTek Cytation3 imaging reader (Winooski, VT) at 595nm. Drug concentrations of GEM (500 µM), MET (400 µM), I3C (250 or 500 µM) were used for the assay. These doses were selected based on a dose-range study. The coefficient of drug interaction (CDI) used to determine whether two drugs are synergistic, additive, or antagonist was calculated with the following equation:
| CID = | (ratio of cell viability for drug A) (ratio of cell viability for drug B) |
| ratio of cell viability for drug combination AB |
RNA extraction and cDNA synthesis
Total RNA were extracted from cells using RNeasy Mini Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. Nucleic acid purities and concentrations were determined using a NanoDrop Spectrophotometer ND1000 (Thermofisher, Grand Island, NY). All RNA samples had A260/A280 ratios of 1.9 to 2.1. The integrity and quality of the RNA were assessed using a Biorad Experion Automated Electrophoresis Station (Hercules, CA), and samples with RNA integrity number (RIN) values between 8 and 10 were utilized. cDNA was synthesized (reverse transcribed polymerase chain reaction (RT-PCR)) from 0.4 µg of high quality RNA (RIN>8) using the Advantage RT-for-PCR Kit according to the manufacturer’s protocol (Clontech Laboratories Inc., Mountain View, CA).
Quantitative real time- polymerase chain reaction (QRT-PCR)
For QRT-PCR, Multiplex master mix solution was obtained from Biorad (Hercules, CA). Primers for SLC 29A1-FAM (hENT1) and β-Actin-VIC were obtained from Life Technologies (Grand Island, NY), and those for SLC29A4 (hENT4) are obtained from Integrated DNA Technologies (Coralville, Iowa). Triplicate experiments were assayed using a Biorad CFX96 C1000 System (Biorad). Delta-Delta Ct (ΔΔCT) calculations were used to determine the change in the relative level of gene expression. β-Actin functioned as the reference gene.
Western blots
After the indicated treatment time, pancreatic cancer cells were washed with PBS, harvested by scraping and lysed in 100 µL RIPA buffer (Thermo Scientific, Grand Island, NY) supplemented with protease inhibitor. Whole cell protein extracts (30 µg) were electrophoresed on TGX stain-free gel (Biorad), and transferred to polyvinylidene difluoride (low fluorescence PVDF; Biorad) membranes. For determining protein expression level of hENT1 and hENT4, membranes were blocked with 3% bovine serum albumin and incubated overnight at 4°C with primary antibody, anti-SLC29A1 (hENT1: rabbit polyclonal; Origene; Rockville, MD) and anti-SLC29A4 (hENT4: mouse monoclonal, Abcam, Cambridge, MA). Membranes were washed and incubated with horseradish peroxidase (HRP) conjugated secondary antibody, goat anti-rabbit IgG or goat anti-mouse IgG (Thermo Scientific). Immunoreactive bands were detected by chemiluminescence using ChemiDoc Touch Imaging System (Biorad). The intensity of each band was quantified by dosimetry using Image LabTM Software (Biorad). Total protein was measured to ensure the protein loading (Supplemental Figure 1).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software Version 6.0 (San Diego, CA). One-way analysis of variance (ANOVA) with Tukey's method to adjust for multiple comparisons was used to determine statistically significance. A p value of <0.05 was considered to be significant.
ResultsTop
MTT assays were performed to investigate the percentage of inhibition of proliferation in pancreatic cancer cell lines when treated with GEM and/or MET or I3C, monotherapy and combination therapy. Treatment with 500 µM GEM resulted in time-dependent inhibition of cell proliferation in MIA PaCa2, PANC1, ASPC1, and SU.86.86 pancreatic cancer cell lines compared to the untreated control. The inhibition of cell proliferation in MIA PaCa2, PANC1, ASPC1, and SU.86.86 pancreatic cancer cell lines at 24 h was 29.5, 50.2, 16.3, and 5.9%, respectively; and at 72 h was 71.6, 81.0, 40.6, and 76.2%, respectively (Figure 1 and 2). Female cell lines, ASPC1 and SU.86.86, were more resistant to GEM treatment than the male cell lines (MIA PaCa2 and PANC1). Treatment with 400 µM MET resulted in decreased inhibition of proliferation in MIA PaCa2, PANC1, ASPC1, and SU.86.86 pancreatic cancer cell lines (24 h: 36.8, 50.2, 8.2, and 0.8%; and 72 h: 23.7, 42.2, 13.8, and 18.6%, respectively) (Figure 1). In male pancreatic cancer cell lines, MIA PaCa2 and PANC1, there was a slight decrease in inhibition of proliferation as treatment time increased; however, there was an increase in inhibition of proliferation in female cell lines, ASPC1 and SU.86.86, as treatment time increased.
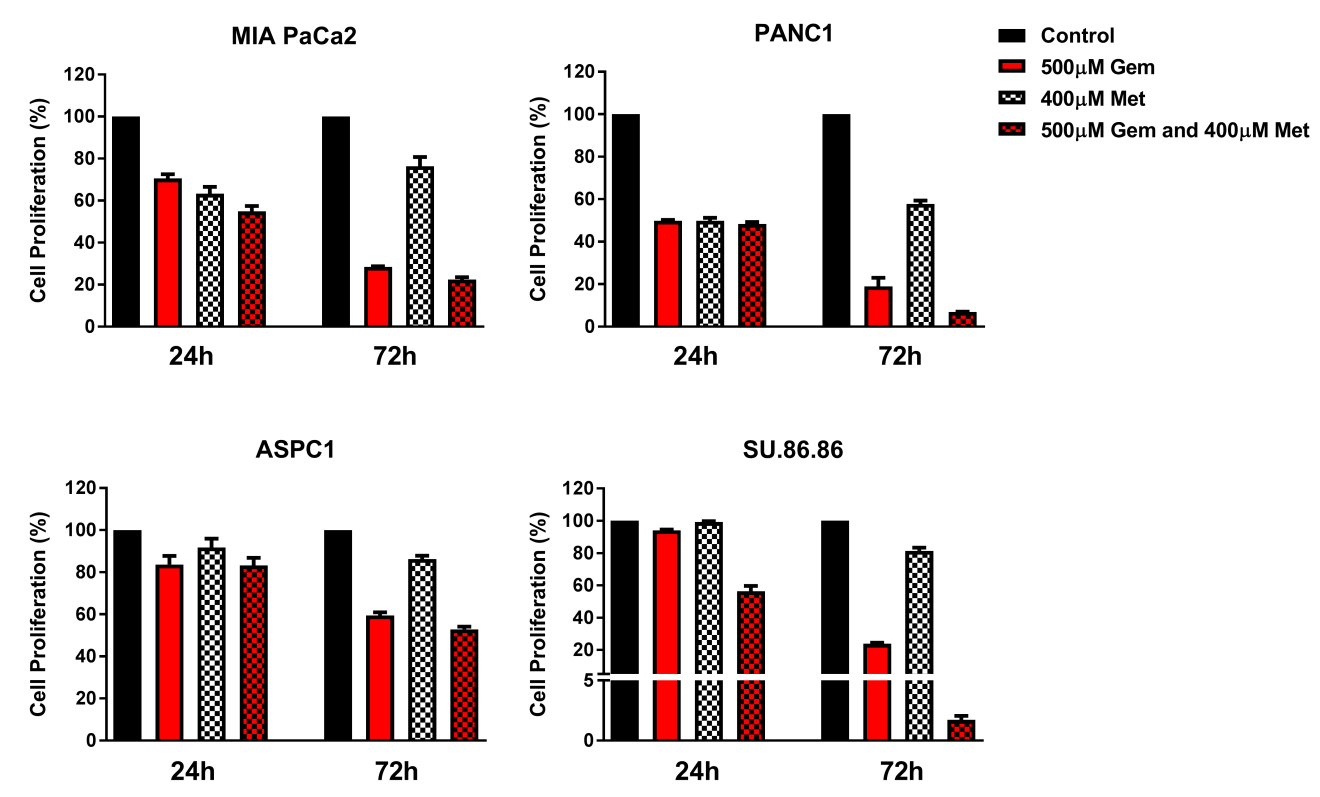
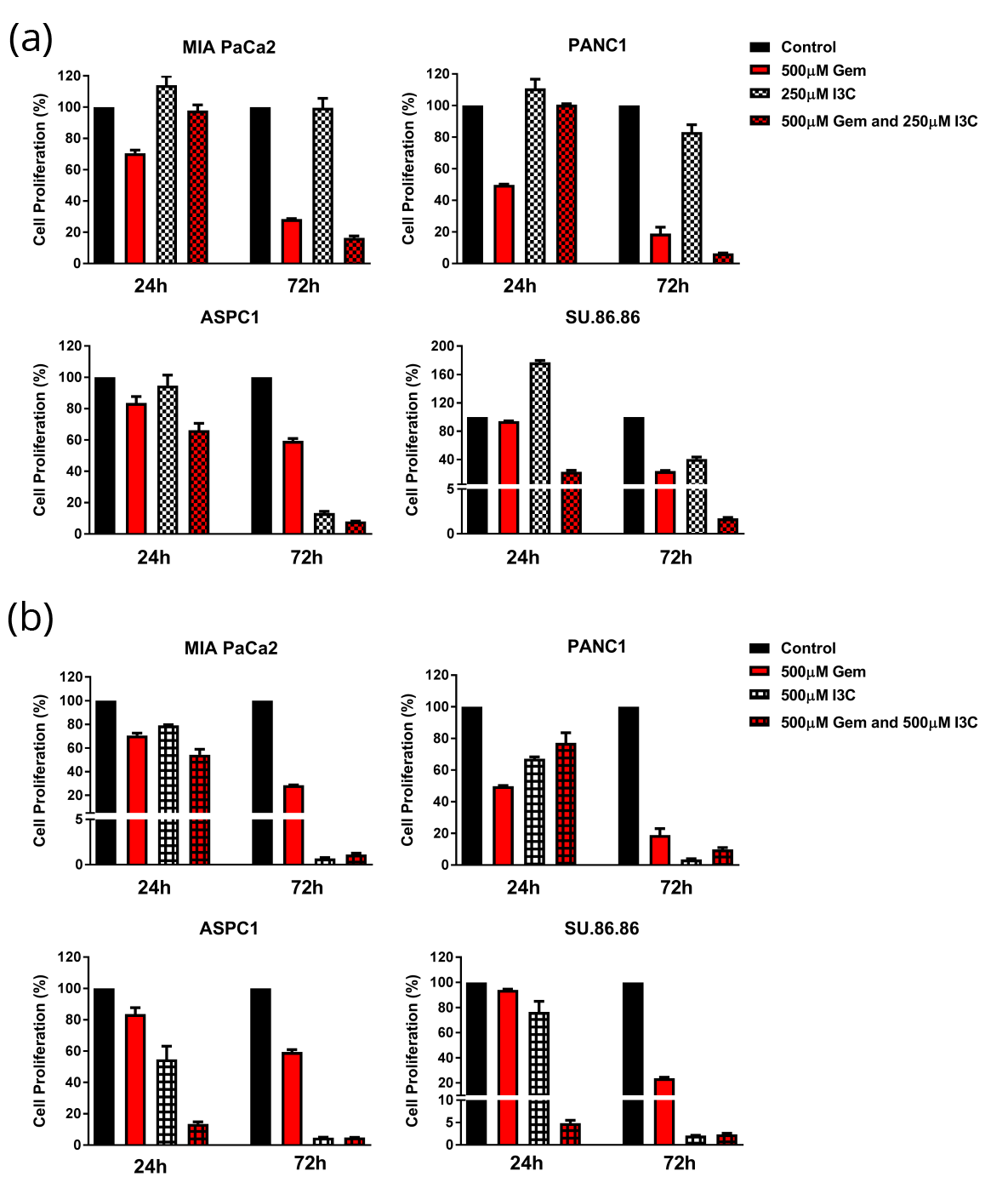
A combination treatment of 500 µM GEM and 400 µM MET resulted in time-dependent inhibition of cell proliferation in MIA PaCa2, PANC1, ASPC1 and SU.86.86 pancreatic cancer cell lines (24 h: 45.2, 51.7, 16.7, and 43.6%; and 72 h: 77.5, 93.0, 47.1, and 98.3%, respectively) (Figure 1). Combination therapy of GEM and MET induced a higher percentage of inhibition of proliferation in pancreatic cancer cells compared to GEM monotherapy at both time points, ranging from 0.4 - 37.7% greater inhibition (Figure 1). However, the summations of cell proliferation inhibition for the individual drugs are higher than the combined drug effect in each of the cell lines with the exception of SU.86.86 (Figure 1). The CDI indicated that treatment with GEM and MET for 24h: ASPC1 and Su.86.86, and 72h: ASPC1, PANC1 and Su.86.86 were synergistic (Table 1). The statistical significance of cell proliferation is found in Table 2. Treatment with I3C resulted in a time- and concentration-dependent increase in inhibition of proliferation of MIA PaCa2, PANC1, ASPC1, and SU.86.86 pancreatic cancer cell lines. After 24 h, the low dose of I3C (250µM) increased cell proliferation in MIA PaCa2, PANC1, and SU.86.86 cells lines by 14.1, 10.8, and 77.1%, respectively, demonstrating that these cells were not affected by the concentration of I3C. In ASPC1 cells, the lower dose of I3C inhibited cell proliferation by 5.2% (Figure 2a). After 72h treatment time, low dose of I3C (250µM) inhibited cell proliferation in MIA PaCa2, PANC1, ASPC1, and SU.86.86 cells lines by 0.3, 16.8, 86.6, and 89.4%, respectively (Figure 2a). By contrast, the high dose of I3C (500µM) resulted in an inhibition of proliferation of 21.0, 32.7, 45.4, and 23.6% at 24h, and cell death 99.3, 96.4, 95.2, and 97.9% at 72h, respectively (Figure 2b).
| Cell line-treatment time | 500µM Gem + 250µM I3c | 500µ M Gem + 500µM I3C | 500µM Gem + 400µM Met |
| MIAPaCa2 - 24h (M) | 1.2 | 1.0 | 1.2 |
| MIAPaCa2 - 72h | 0.6 | 5.7 | 1.0 |
| ASPC1 - 24h (F) | 0.8 | 0.3 | 0.8 |
| ASPC1 - 72h | 1.0 | 1.7 | 0.4 |
| PANC1 - 24h (M) | 1.8 | 2.3 | 1.9 |
| PANC1 - 72h | 0.4 | 14.6 | 0.6 |
| SU.86.86 - 24h (F) | 0.1 | 0.1 | 0.8 |
| Su.86.86 - 72h | 0.2 | 4.8 | 0.1 |
Abbreviations: M: male; F: female; CDI: =1.0 (additive), >1.0 (antagonist), <1.0 (synergist).
| Treatment | MIAPaCa2 (Male) | |
| 24 h | 72 h | |
| Control vs. 400 µM Met | < 0.0001 | 0.0002 |
| Control vs. 500 µM Gem | 0.0003 | < 0.0001 |
| Control vs. 250 µM I3C | 0.1752 | > 0.9999 |
| Control vs. 500 µM I3C | 0.0286 | < 0.0001 |
| Control vs. 500 µM Gem and 400 µM Met | < 0.0001 | < 0.0001 |
| Control vs. 500 µM Gem and 250 µM I3C | > 0.9999 | < 0.0001 |
| Control vs. 500 µM Gem and 500 µM I3C | < 0.0001 | < 0.0001 |
| 400 µM Met vs. 400 µM Met and 500 µM Gem | 0.7663 | < 0.0001 |
| 500 µM Gem vs. 500 µM Gem and 400 µM Met | 0.0961 | 0.8553 |
| 500 µM Gem vs. 500 µMGem and 250 µM I3C | 0.0008 | 0.1089 |
| 500 µM Gem vs. 500 µM Gem and 500 µM I3C | 0.0800 | < 0.0001 |
| 250 µM I3C vs. 500 µM I3C | 0.0001 | < 0.0001 |
| 250 µM I3C vs. 500 µM Gem and 250 µM I3C | 0.0805 | < 0.0001 |
| 500 µM I3C vs. 500 µM Gem and 500 µM I3C | 0.0071 | > 0.9999 |
| 500 µM Gem and 500 µM I3C vs. 500 µMGem and 250 µM I3C | < 0.0001 | 0.0192 |
| Treatment | ASPC1 (Female) | |
| 24 h | 72 h | |
| Control vs. 400 µM Met | 0.922 | < 0.0001 |
| Control vs. 500 µM Gem | 0.2255 | < 0.0001 |
| Control vs. 250 µM I3C | 0.9957 | < 0.0001 |
| Control vs. 500 µM I3C | 0.9986 | < 0.0001 |
| Control vs. 500 µM Gem and 400 µM Met | 0.0005 | < 0.0001 |
| Control vs. 500 µM Gem and 250 µM I3C | 0.2041 | < 0.0001 |
| Control vs. 500 µM Gem and 500 µM I3C | < 0.0001 | < 0.0001 |
| 400 µM Met vs. 400 µM Met and 500 µM Gem | 0.0106 | < 0.0001 |
| 500 µM Gem vs. 500 µM Gem and 400 µM Met | 0.1649 | < 0.0001 |
| 500 µM Gem vs. 500 µMGem and 250 µM I3C | > 0.9999 | 0.0017 |
| 500 µM Gem vs. 500 µM Gem and 500 µM I3C | 0.0015 | < 0.0001 |
| 250 µM I3C vs. 500 µM I3C | > 0.9999 | > 0.9999 |
| 250 µM I3C vs. 500 µM Gem and 250 µM I3C | 0.6581 | < 0.0001 |
| 500 µM I3C vs. 500 µM Gem and 500 µM I3C | < 0.0001 | 0.0002 |
| 500 µM Gem and 500 µM I3C vs. 500 µMGem and 250 µM I3C | 0.0017 | < 0.0001 |
| Treatment | PANC1 (Male) | |
| 24 h | 72 h | |
| Control vs. 400 µM Met | < 0.0001 | < 0.0001 |
| Control vs. 500 µM Gem | < 0.0001 | < 0.0001 |
| Control vs. 250 µM I3C | 0.1745 | 0.0006 |
| Control vs. 500 µM I3C | < 0.0001 | < 0.0001 |
| Control vs. 500 µM Gem and 400 µM Met | < 0.0001 | < 0.0001 |
| Control vs. 500 µM Gem and 250 µM I3C | > 0.9999 | < 0.0001 |
| Control vs. 500 µM Gem and 500 µM I3C | 0.0012 | < 0.0001 |
| 400 µM Met vs. 400 µM Met and 500 µM Gem | > 0.9999 | < 0.0001 |
| 500 µM Gem vs. 500 µM Gem and 400 µM Met | > 0.9999 | 0.0187 |
| 500 µM Gem vs. 500 µMGem and 250 µM I3C | < 0.0001 | 0.0131 |
| 500 µM Gem vs. 500 µM Gem and 500 µM I3C | 0.0001 | 0.1247 |
| 250 µM I3C vs. 500 µM I3C | < 0.0001 | < 0.0001 |
| 250 µM I3C vs. 500 µM Gem and 250 µM I3C | 0.2189 | < 0.0001 |
| 500 µM I3C vs. 500 µM Gem and 500 µM I3C | 0.4859 | 0.53 |
| 500 µM Gem and 500 µM I3C vs. 500 µMGem and 250 µM I3C | 0.0009 | 0.9745 |
| Treatment | SU.86.86 (Female) | |
| 24 h | 72 h | |
| Control vs. 400 µM Met | > 0.9999 | < 0.0001 |
| Control vs. 500 µM Gem | 0.8911 | < 0.0001 |
| Control vs. 250 µM I3C | < 0.0001 | < 0.0001 |
| Control vs. 500 µM I3C | < 0.0001 | < 0.0001 |
| Control vs. 500 µM Gem and 400 µM Met | < 0.0001 | < 0.0001 |
| Control vs. 500 µM Gem and 250 µM I3C | 0.0123 | < 0.0001 |
| Control vs. 500 µM Gem and 500 µM I3C | < 0.0001 | < 0.0001 |
| 400 µM Met vs. 400 µM Met and 500 µM Gem | < 0.0001 | < 0.0001 |
| 500 µM Gem vs. 500 µM Gem and 400 µM Met | < 0.0001 | < 0.0001 |
| 500 µM Gem vs. 500 µMGem and 250 µM I3C | 0.2215 | 0.9998 |
| 500 µM Gem vs. 500 µM Gem and 500 µM I3C | < 0.0001 | < 0.0001 |
| 250 µM I3C vs. 500 µM I3C | 0.9996 | < 0.0001 |
| 250 µM I3C vs. 500 µM Gem and 250 µM I3C | < 0.0001 | < 0.0001 |
| 500 µM I3C vs. 500 µM Gem and 500 µM I3C | < 0.0001 | < 0.0001 |
| 500 µM Gem and 500 µM I3C vs. 500 µMGem and 250 µM I3C | < 0.0001 | < 0.0001 |
Combined GEM and I3C therapy (both low and high dose) induced a higher percentage of inhibition of proliferation in female pancreatic cancer cells (ASPC1 and SU.86.86) compared to GEM monotherapy at both time points, ranging from 17.5%-89.2% (Figure 2). However, at 24h treatment time, GEM monotherapy inhibited cell proliferation at a higher rate in MIA PaCa2 compared to combination therapy with low dose of I3C, and in PANC1 compared to combination therapy with either dose of I3C (Figure 2). At 72h, combined therapy of GEM and I3C (both low and high dose) induced a higher percentage of inhibition of proliferation in male pancreatic cancer cells (MIA PaCa2 and PANC1) compared to GEM monotherapy at both time points, ranging from 9.1%-27.2% (Figure 2). The combined drug effects of GEM and I3C are higher than summations of cell proliferation inhibition for the two drugs for the following cell lines (I3C dose and treatment time): APSC1 and SU.86.86 (24h, both I3C doses), and MIA PaCa2 (72h, low dose I3C) (Figure 2).
The CDI indicated that GEM and low dose I3C (250µM) were found be synergistic in MIA PaCa2 (72h), ASPC1 (24h), PANC1 (72h), and SU.86.86 (24h and 72h); and GEM and high dose I3C (500µM) were found be synergistic in ASPC1 (24h), and SU.86.86 (24h) (Table 1). The statistical significance of cell proliferation is found in Table 2a-d.
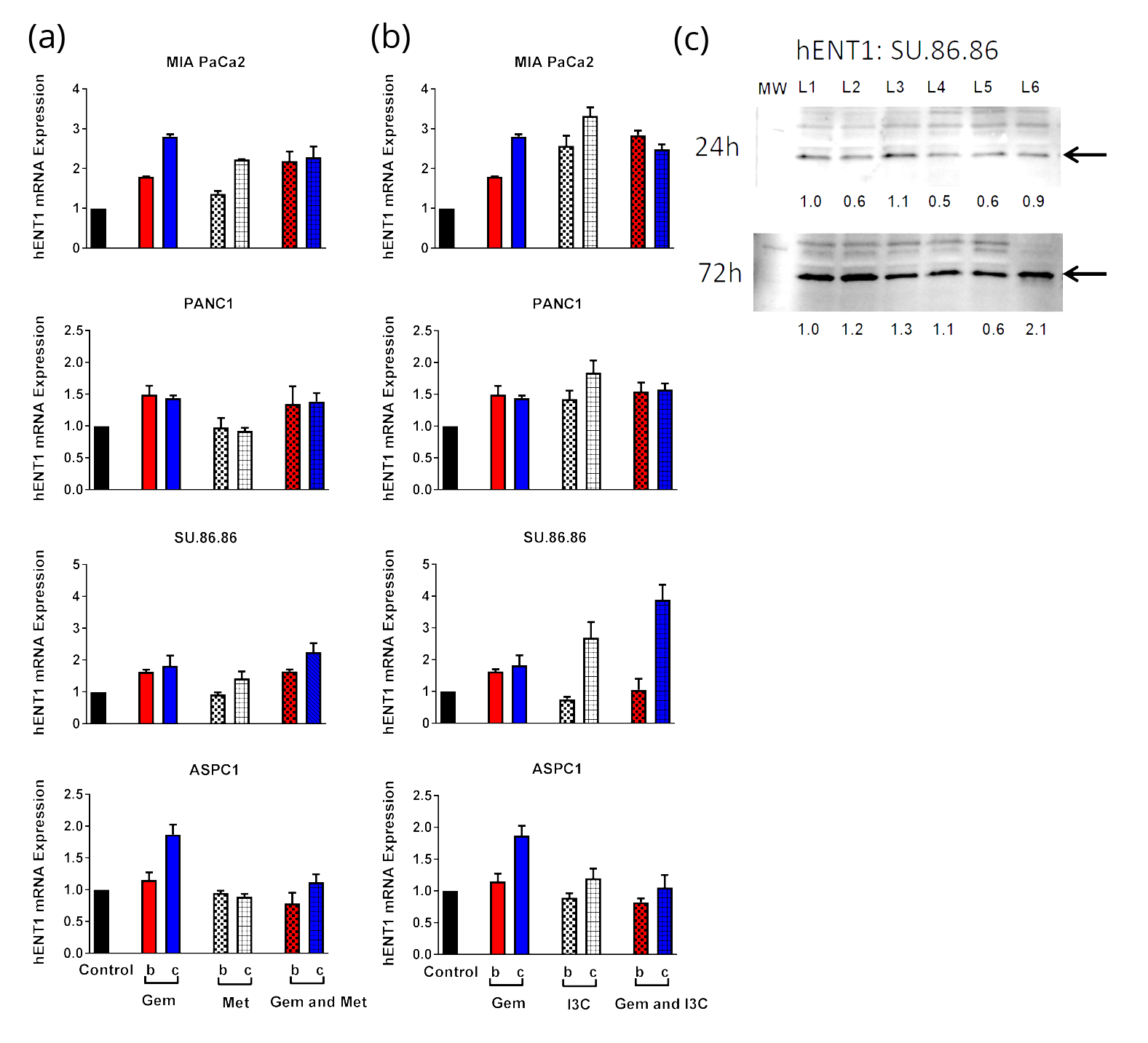
The expression levels of two drug transporter genes, hENT1 and hENT4, were investigated in pancreatic cancer cell lines before and after drug treatment with 500µM GEM and/or 400µM MET or 250µMI3C. On average MET induced lower mRNA expression of hENT1 in pancreatic cancer cells compared to GEM. The highest mRNA expression of hENT1 induced by MET was found in MIA PaCa2 cells (72h) (Figure 3a). Combined therapy of GEM and MET was found to enhance the hENT1 mRNA expression only in SU.86.86 cells after 72h treatment time by 10% compared to the summation of percent increase induced by the individual drugs (Figure 3a). Protein analysis did not confirm this finding (Figure 3c). 250µM I3C induced hENT1 mRNA expression in all cell lines except ASPC1 in a time-dependent manner (Figure 3b). Indole-3-carbinaol induced higher mRNA expression of hENT1 in MIA PaCa2 (24h and 72h), PANC1 (72h) and SU.86.86 (72h) pancreatic cancer cells compared to GEM. Combined therapy of GEM and 250µM I3C was found to enhance the hENT1 mRNA expression only in SU.86.86 cells after 72h treatment time by 40% compared to the summation of percent increase induced by the individual drugs (Figure 3b). Protein analysis confirmed this finding in SU.86.86 cells treated with GEM and 250µM I3C for 72h (2.1-fold, Figure 3c). On average GEM induced higher mRNA expression levels of hENT4 compared to MET (Figure 4b).
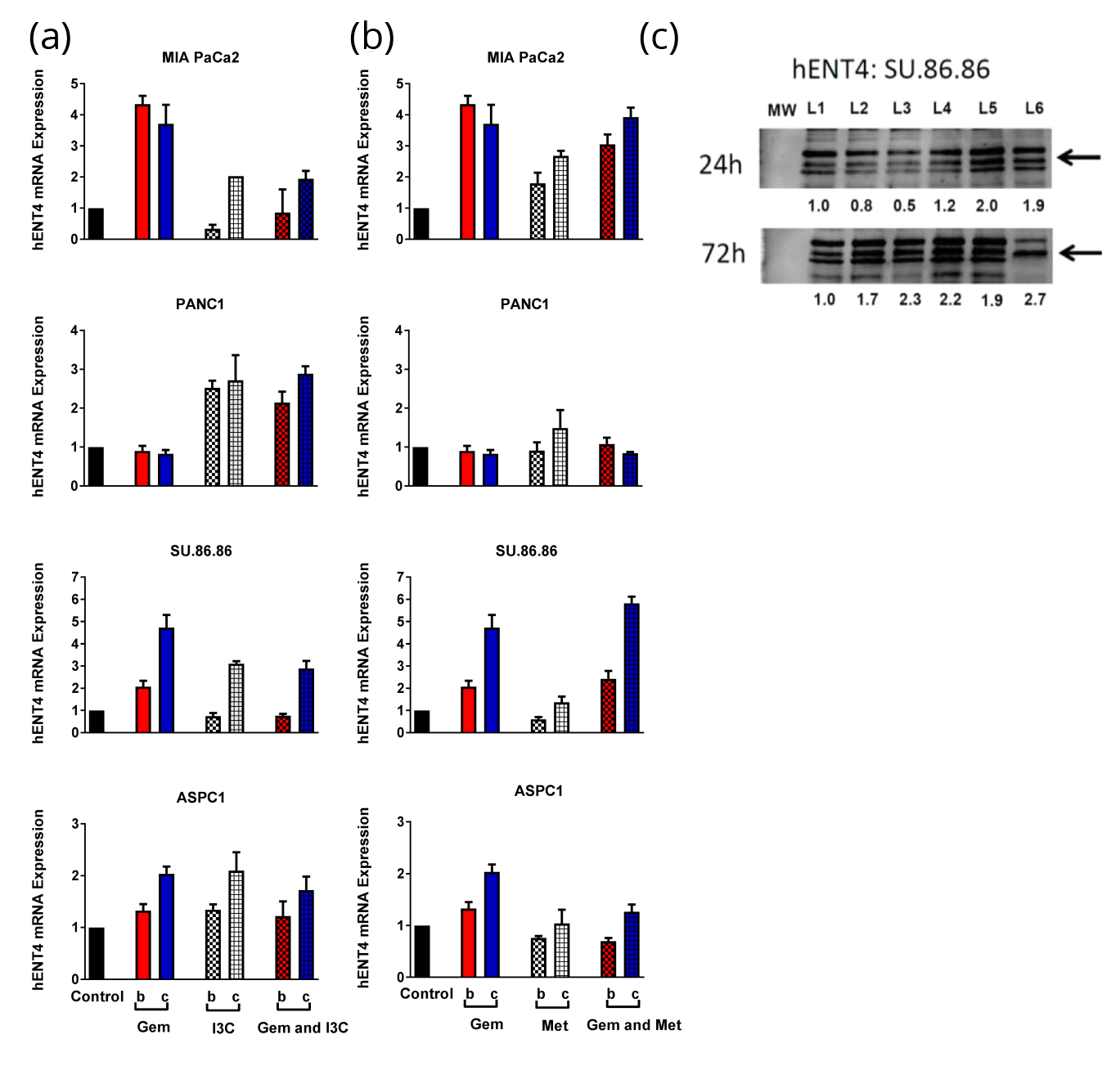
Combined treatment of GEM and MET enhanced hENT4 mRNA expression in SU.86.86 pancreatic cancer cell lines after 24h and 72h treatment time by 70% and 80% compared to the summation of percent increase induced by the individual drugs (Figure 4a). Protein analysis confirmed this finding in SU.86.86 cells treated with GEM and MET for 24h and 72h (2.0- and 1.9-fold, respectively; Figure 3c). Combined treatment of GEM and 250M I3C did not enhance hENT4 mRNA expression in pancreatic cancer cell lines after 24h and 72h treatment time compared to the summation of the individual drugs. The statistical significance of mRNA expression analysis is found in Table 3 (a and b).
| MIA PaCa2 (M) | PANC1 (M) | ASPC1 (F) | Su.86.86 (F) | ||||||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | ||
| hENT1 | Control vs. 400 µM Met | 0.4668 | 0.0058 | > 0.9999 | 0.9857 | 0.9989 | 0.9886 | 0.9989 | 0.8658 |
| Control vs. 500 µM Gem | 0.0386 | 0.0001 | 0.3136 | 0.0393 | 0.8346 | 0.0051 | 0.1006 | 0.3301 | |
| Control vs. 250 µM I3C | 0.0003 | < 0.0001 | 0.4596 | 0.001 | 0.9592 | 0.8748 | 0.8493 | 0.0238 | |
| Control vs. 500 µM Gem + 400 µM Met | 0.0012 | 0.0018 | 0.6528 | 0.0784 | 0.5772 | 0.9843 | 0.0891 | 0.0669 | |
| Control vs. 500 µM Gem + 250 µM I3C | < 0.0001 | 0.0015 | 0.2265 | 0.0155 | 0.7515 | 0.9995 | 0.9999 | 0.0005 | |
| 400 µM Met vs. 500 µM Gem + 400 µM Met | 0.017 | 0.9999 | 0.5948 | 0.03 | 0.7804 | 0.8025 | 0.0485 | 0.3284 | |
| 500 µM Gem vs. 500 µM Gem + 400 µM Met | 0.5181 | 0.2763 | 0.9846 | 0.9968 | 0.1152 | 0.015 | > 0.9999 | 0.8642 | |
| 500 µM Gem vs. 500 µM Gem + 250 µM I3C | 0.0076 | 0.7948 | 0.9999 | 0.9074 | 0.1864 | 0.0084 | 0.1469 | 0.0067 | |
| 250 µM I3C vs. 500 µM Gem + 250 µM I3C | 0.828 | 0.06 | 0.9934 | 0.5131 | 0.9932 | 0.9642 | 0.7308 | 0.1968 | |
| 500 µM Gem + 400 µMet vs. 500 µM Gem + 250 µM I3C | 0.0647 | 0.963 | 0.9446 | 0.7228 | 0.9995 | 0.9992 | 0.1306 | 0.0294 | |
Abbreviations: M: male; F: female.
| MIA PaCa2 (M) | PANC1 (M) | ASPC1 (F) | Su.86.86 (F) | ||||||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | ||
| hENT4 | Control vs. 400 µM Met | 0.6799 | 0.0297 | 0.9992 | 0.7963 | 0.5714 | > 0.9999 | 0.845 | 0.9623 |
| Control vs. 500 µM Gem | 0.0005 | 0.0009 | 0.9987 | 0.9969 | 0.2757 | 0.1174 | 0.054 | 0.0001 | |
| Control vs. 250 µM I3C | 0.8146 | 0.3818 | 0.0008 | 0.0236 | 0.3263 | 0.0503 | 0.97 | 0.0197 | |
| Control vs. 500 µM Gem + 400 µM Met | 0.023 | 0.0005 | 0.9995 | 0.9982 | 0.343 | 0.9561 | 0.0106 | < 0.0001 | |
| Control vs. 500 µM Gem + 250 µM I3C | 0.9998 | 0.3561 | 0.0081 | 0.0132 | 0.725 | 0.296 | 0.9796 | 0.0376 | |
| 400 µM Met vs. 500 µM Gem + 400 µM Met | 0.2527 | 0.1383 | 0.9843 | 0.5779 | 0.9968 | 0.9776 | 0.0044 | < 0.0001 | |
| 500 µM Gem vs. 500 µM Gem + 400 µM Met | 0.2246 | 0.9962 | 0.9798 | > 0.9999 | 0.0117 | 0.3396 | 0.8411 | 0.2518 | |
| 500 µM Gem vs. 500 µM Gem + 250 µM I3C | 0.0003 | 0.0217 | 0.0043 | 0.0073 | 0.983 | 0.946 | 0.0346 | 0.042 | |
| 250 µM I3C vs. 500 µM Gem + 250 µM I3C | 0.9214 | > 0.9999 | 0.6886 | 0.999 | 0.9772 | 0.8407 | > 0.9999 | 0.9984 | |
| 500 µM Gem + 400 µMet vs. 500 µM Gem + 250 µM I3C | 0.0147 | 0.0102 | 0.0136 | 0.0078 | 0.0638 | 0.7228 | 0.0082 | 0.002 | |
Abbreviations: M: male; F: female.
DiscussionTop
GEM monotherapy has been the standard of treatment for metastatic pancreatic cancer patients for several years; however, the majority of patients treated with GEM eventually become resistant to the drug. Pancreatic cancer cell lines possess high intrinsic and acquired chemoresistance. Pang et al. showed that polymorphisms in ABCB1 in pancreatic cancer cell lines affected GEM efficacy, where MIA PaCa2 cells exhibit a ABCB12677TT polymorphism, and are more sensitive to GEM than PANC1, SU.86.86 and ASPC1 [7]. Therefore, studies have investigated the use of GEM in combination therapy for the re-sensitization and treatment of pancreatic cancer. Combination therapy of GEM and erlotinib was shown to increase the median survival of patients with metastatic pancreatic cancer by two weeks [28-30]. However, this treatment had substantial side effects, as well as high cost compared to the modest survival benefits [30, 31]. The multidrug combination of leucovorin, fluorouracil, irinotecan, and oxaliplatin also known as folfirinox, was found to increase the median survival of 4.3 months. Despite the improved median of survival, these treatments were found to have significantly higher levels of toxicity [32]. One of the more recent combination therapies is GEM plus nab-paclitaxel. This treatment increased the median survival by 1.87 months, increased the overall survival at 1 and 2 years, and had reasonable adverse effects [33]. Several of these combinations therapies are currently being used for patients with good performance status based on their Eastern Cooperative Oncology Group (ECOG) performance status [30]. For metastatic pancreatic cancer patients with poor performance status, GEM (monotherapy) remains the recommended treatment. Therefore, despite advances in chemotherapeutics and the understanding of the biology of pancreatic cancer, there has been limited progress in therapy options for metastatic pancreatic cancer, and the need to evaluate other potential therapeutics continues. In the present study, we evaluated the combined drug effects of GEM and/or MET or I3C in pancreatic cancer cells of males and females. Several pancreatic cancer cell lines were examined for cell viability, drug interaction (summation and synergistic effect), and modulation of critical transporters, hENT1 and hENT4, expression when treated with GEM, MET, I3C in mono or GEM combination therapy for 24h and 72h. The female cell lines, ASPC1 and SU86.86 were found to exhibit synergistic drug behavior when treated with GEM and MET at both time points.
I3C, a compound found in Brassica vegetables such as broccoli, has been found to be well tolerated in high doses in both animals and humans [13, 34], and to be a promising nontoxic chemopreventive agent [9]. While potential use in humans has been shown to be feasible, concerns remain as to its tendency to induce activating enzymes, such as cytochrome P450s, and its promoting activity under uncertain experimental conditions [66]. Under certain experimental conditions, I3C can act as a tumor promoter in rodents [35, 36]. In this study we observed that a low dose of I3C (250µM) induced an increase in cell proliferation of pancreatic cancer cells after 24 h of treatment. Despite this concern, several studies have shown that I3C exhibit its anticancer properties in various cancer cells [37-39]. I3C has inhibited tumorigenesis in breast, liver, lung, cervix, and gastrointestinal tract in different animal models [13, 14, 40-42]. Inhibition of tumorigenesis using I3C occurs via different mechanisms [43, 44]. The current study demonstrated that treatment with I3C resulted in a time- and concentration-dependent increase in inhibition of proliferation of MIA PaCa2, PANC1, ASPC1, and SU.86.86 pancreatic cancer cell lines, where male pancreatic cancer cells, MIA PaCa2 and PANC1, treated with low dose of I3C (250µM) exhibited the lowest cytotoxicity levels.
As part of this study, we were also interested in whether members of the hENT family of transporters, namely hENT1 and hENT4, play a role in the increased cytotoxicity of GEM combined therapies. To evaluate the role of hENTs in GEM combined therapies, we compared the current study to a previous study performed by our lab. The most noticeable difference between the previous study and the current study is the method of treatment. In the previous study, cells were initially pre-treated with I3C for 24 h and subsequently treated with both I3C and GEM [6]. Based on the observations from that study, pre-treatment with I3C followed by treatment with I3C and GEM not only significantly decreased pancreatic cancer cell viability by enhancing the efficacy of GEM but required less drug concentration to achieve this outcome [6]. The study also demonstrated I3C enhancement of GEM cytotoxicity in pancreatic cancer through the upregulation of hENT1, which is involved in the transport of GEM [6]. In the current study the pancreatic cancer cells were not pre-treated with I3C before subsequent combination drug therapy treatment. While both studies demonstrated that treatment with I3C and GEM could significantly decrease cell viability in pancreatic cancer cell line compared to GEM-only treatment, there was a difference in their regulation of hENT1. Pancreatic cancer cells treated with GEM and I3C in the current study did not have significantly upregulated hENT1 expression, with the exception of SU.86.86 cells after 72h treatment. GEM and I3C combined therapy also did not upregulated the expression of hENT4. This difference in treatment methods could account for differences in drug responses and hENT expression levels. We postulate that pre-treatment with I3C followed by subsequent combined therapy with GEM better sensitizes the pancreatic cancer cells to GEM therapy through one of several mechanisms. This may occur through several biological mechanisms, such as, the inhibition of cell proliferation through modulation of various proteins involved in induction of pro-apoptotic proteins, inhibition of anti-apoptotic proteins, the inhibition of signaling pathways involved in cell survival, the induction of cytochrome P450, and cell cycle regulation [12, 43]. In breast cancer it has previously been shown that I3C synergizes with tamoxifen to more stringently induce G1 cell cycle arrest through the additive inhibition of CDK2 kinase activity [45]. I3C has also been shown to down regulate of miR-21, which overexpression is linked to chemoresistance in pancreatic cancer [46], reactivate p16INK4a tumor suppression gene through hypomethylation of the promoter [47], and a previous study performed by our lab showed that pre-treating with I3C enhanced the expression of hENT1 [6]. Consequently, pre-treatment of pancreatic cancer cells with I3C may potentially sensitize pancreatic cancer cells through many of the above biological mechanisms which increase GEM efficacy.
Another therapeutic agent used in combination with GEM is MET. MET is a well-known inexpensive anti-diabetic agent that has been shown to have anticancer properties [48-52]. Studies have illustrated that the mortality rate from cancer was lower with diabetics patients taking MET (pre-treatment with MET) as part of their anti-diabetic mediation compared to those not taking MET and to other insulin therapies [22, 53-56]. In this study, we demonstrate that MET monotherapy inhibition of cell proliferation in pancreatic cancer cells was cell line specific. The inhibition of cell proliferation in cancer cells using MET is supported by several studies, where MET has been shown to exert in vitro inhibition of proliferation in prostate, ovarian, colorectal and breast cancer cells [56-61], to significantly prevent pancreatic cancer development in hamsters [62], and to target pancreatic cancer stem cells [23]. In recent years, there has been an increase in the number of clinical trials investigating the involvement of MET in cancer. Most of these trials involve MET in combination therapy. In a double-blind, placebo-controlled phase II clinical trial of MET in pancreatic cancer treatment with a survival endpoint, there was no advantage for the addition of MET to erlotniab and GEM [26]. The use of MET and paclitaxel as a second line treatment for advanced pancreatic cancer was found to be poorly tolerated and the primary end point (disease control rate) was not met [63]. Some of the limitations noted in these clinical studies are self-reporting from diabetic patients, misclassification of patients, incomplete medication records, time-related bias studies, and small population size [19, 21, 53, 64, 65]. Therefore, studies interested in whether MET sensitize pancreatic cancer cells to GEM and enhances the capacity of GEM to inhibit invasion and proliferation of pancreatic cancer cells are continuing to be evaluated.
ConclusionTop
In this study, pancreatic cancer cells treated with GEM and MET demonstrated time-dependent decrease in cell viability. Overall the female cell lines, ASPC1 and SU.86.86, were found to exhibit synergistic drug behavior when treated with GEM and MET at both time points. Overall the efficacy of MET in combination with GEM treatment was cell line specific and may also be dependent on the method of treatment. MET may potentially have a similar drug response to I3C, where the treatment method may influence the cytotoxicity and hENT expression in pancreatic cancer cells. We propose that pre-treatment with MET may enhance cytotoxicity and hENT expression in pancreatic cells; however, further studies are needed to verify this hypothesis. Combination treatment of GEM and MET or I3C has the potential to enhance the ability of GEM and inhibit cell proliferation of pancreatic cancer cells through members of the hENT family of transporters. However, genetic variability in hENT drug transporters is also an important factor in gemcitabine response. In addition, MET targets a number of signaling pathways that play roles in cell proliferation and angiogenesis, such mTOR. The overall effect of GEM combination, therapy in pancreatic cancer cells may be cell line- and treatment method-specific, where pre-treatment with I3C or MET better sensitizes pancreatic cancer cells to GEM. To better understand the impact of treatment method, a detailed time-, concentration- and treatment method-dependent matched study will have to be performed to confirm these initial findings.
Competing interests
The authors declare that they have no competing interests.
Funding
This study was funded by the FDA National Center for Toxicological Research.
Acknowledgement
SJ was supported by an appointment to the Postgraduate Research Program at the National Center for Toxicological Research administered by the Oak Ridge Institute for Science Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration. The views presented in this report do not necessarily reflect those of the US Food and Drug Administration.
Supplementary dataTop
ReferencesTop
[1]Cancer Facts & Figures 2016. American Cancer Society.Article
[2]Cid-Arregui A, Juarez V. Perspectives in the treatment of pancreatic adenocarcinoma. World J Gastroenterol. 2015; 21(31):9297–9316.Article Pubmed
[3]Tempero MA, Arnoletti JP, Behrman SW, Ben-Josef E, Benson AB, et al. Pancreatic adenocarcinoma, version 2.2012: Featured updates to the NCCN guidelines. J Natl Compr Canc Netw. 2012; 10(6):703–713.Article Pubmed
[4]Tanaka M, Okazaki T, Suzuki H, Abbruzzese JL, Li D. Association of multi-drug resistance gene polymorphisms with pancreatic cancer outcome. Cancer. 2011; 117(4):744–7451.Article Pubmed
[5]Amrutkar M, Gladhaug IP. Pancreatic cancer chemoresistance to gemcitabine. Cancers. 2017; 9(11):157.Article Pubmed
[6]Wang H, Word BR, Lyn-Cook BD. Enhanced efficacy of gemcitabine by indole-3-carbinol in pancreatic cell lines: The role of human equilibrative nucleoside transporter 1. Anticancer Res. 2011; 31(10):3171–3180.Article Pubmed
[7]Pang L, Word B, Xu J, Wang H, Hammons G, et al. ATP-binding cassette genes genotype and expression: A potential association with pancreatic cancer development and chemoresistance? Gastroenterol Res Pract. 2014; 2014:414931.Article Pubmed
[8]Abdelmageed MM, El-Naga RN, El-Demerdash E, Elmazar MM. Indole-3- carbinol enhances sorafenib cytotoxicity in hepatocellular carcinoma cells: A mechanistic study. Sci Rep. 2016; 6:32733.Article Pubmed
[9]He YH, Schut HA. Inhibition of DNA adduct formation of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and 2-amino-3-methylimidazo[4,5-f]quinoline by dietary indole-3-carbinol in female rats. J Biochem Mol Toxicol. 1999; 13(5):239–247.Article Pubmed
[10]Bell MC, Crowley-Nowick P, Bradlow HL, Sepkovic DW, Schmidt-Grimminger D, et al. Placebo-controlled trial of indole-3-carbinol in the treatment of CIN. Gynecol Oncol. 2000; 78(2):123–129.Article Pubmed
[11]Rosen CA, Bryson PC. Indole-3-carbinol for recurrent respiratory papillomatosis: long-term results. J Voice. 2004; 18(2):248–253.Article Pubmed
[12]Aronchik I, Bjeldanes LF, Firestone GL. Direct inhibition of elastase activity by indole-3-carbinol triggers a CD40-TRAF regulatory cascade that disrupts NF-kappaB transcriptional activity in human breast cancer cells. Cancer Res. 2010; 70(12):4961–4971.Article Pubmed
[13]Bradlow HL, Michnovicz J, Telang NT, Osborne MP. Effects of dietary indole-3-carbinol on estradiol metabolism and spontaneous mammary tumors in mice. Carcinogenesis. 1991; 12(9):1571–1574.Article Pubmed
[14]Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001; 20(23):2927–2936.Article Pubmed
[15]Ashida R, Nakata B, Shigekawa M, Mizuno N, Sawaki A, et al. Gemcitabine sensitivity-related mRNA expression in endoscopic ultrasound-guided fine-needle aspiration biopsy of unresectable pancreatic cancer. J Exp Clin Cancer Res. 2009; 28:83.Article Pubmed
[16]Eto K, Kawakami H, Kuwatani M, Kudo T, Abe Y, et al. Human equilibrative nucleoside transporter 1 and Notch3 can predict gemcitabine effects in patients with unresectable pancreatic cancer. Br J Cancer. 2013; 108(7):1488–1494.Article Pubmed
[17]Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014; 10(3):143–156.Article Pubmed
[18]Zhou M, Xia L, Wang J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab Dispos. 2007; 35(10):1956–1962.Article Pubmed
[19]Sadeghi N, Abbruzzese JL, Yeung SC, Hassan M, Li D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin Cancer Res. 2012; 18(10):2905–2912.Article Pubmed
[20]Suissa S, Azoulay L. Metformin and cancer: Mounting evidence against an association. Diabetes Care. 2014; 37(7):1786–1788.Article Pubmed
[21]Wang Z, Lai ST, Xie L, Zhao JD, Ma NY, et al. Metformin is associated with reduced risk of pancreatic cancer in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Res Clin Pract. 2014; 106(1):19–26.Article Pubmed
[22]Yang YX. Do diabetes drugs modify the risk of pancreatic cancer? Gastroenterology. 2009; 137(2):412–415.Article Pubmed
[23]Lonardo E, Cioffi M, Sancho P, Sanchez-Ripoll Y, Trabulo SM, et al. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PloS one. 2013; 8(10):e76518.Article Pubmed
[24]Chai X, Chu H, Yang X, Meng Y, Shi P, et al. Metformin increases sensitivity of pancreatic cancer cells to gemcitabine by reducing CD133+ cell populations and suppressing ERK/P70S6K signaling. Sci Rep. 2015; 5:14404.Article Pubmed
[25]Fasih A, Elbaz HA, Huttemann M, Konski AA, Zielske SP. Radiosensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat Res. 2014; 182(1):50–59.Article Pubmed
[26]Kordes S, Pollak MN, Zwinderman AH, Mathot RA, Weterman MJ, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015; 16(7):839–847.Article Pubmed
[27]Lipner MB, Marayati R, Deng Y, Wang X, Raftery L, et al. Metformin treatment does not inhibit growth of pancreatic cancer patient-derived xenografts. PloS one. 2016; 11(1):e0147113.Article Pubmed
[28]Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J Clin Oncol. 1997; 15(6):2403–2413.Article Pubmed
[29]Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007; 25(15):1960–1966.Article Pubmed
[30]Thota R, Pauff JM, Berlin JD. Treatment of metastatic pancreatic adenocarcinoma: A review. Oncology (Williston Park). 2014; 28(1):70–74.Article Pubmed
[31]Miksad RA, Schnipper L, Goldstein M. Does a statistically significant survival benefit of erlotinib plus gemcitabine for advanced pancreatic cancer translate into clinical significance and value? J Clin Oncol. 2007; 25(28):4506–4507.Article Pubmed
[32]Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364(19):1817–1825.Article Pubmed
[33]Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013; 369(18):1691–1703.Article Pubmed
[34]Grubbs CJ, Steele VE, Casebolt T, Juliana MM, Eto I, et al. Chemoprevention of chemically-induced mammary carcinogenesis by indole-3-carbinol. Anticancer Res. 1995; 15(3):709–716.Pubmed
[35]Kim DJ, Han BS, Ahn B, Hasegawa R, Shirai T, et al. Enhancement by indole-3-carbinol of liver and thyroid gland neoplastic development in a rat medium-term multiorgan carcinogenesis model. Carcinogenesis. 1997; 18(2):377–381.Article Pubmed
[36]Pence BC, Buddingh F, Yang SP. Multiple dietary factors in the enhancement of dimethylhydrazine carcinogenesis: main effect of indole-3-carbinol. J Natl Cancer Inst. 1986; 77(1):269–276.Pubmed
[37]Frydoonfar HR, McGrath DR, Spigelman AD. Inhibition of proliferation of a colon cancer cell line by indole-3-carbinol. Colorectal Dis. 2002; 4(3):205–207.Article Pubmed
[38]Nachshon-Kedmi M, Yannai S, Haj A, Fares FA. Indole-3-carbinol and 3,3'-diindolylmethane induce apoptosis in human prostate cancer cells. Food Chem Toxicol. 2003; 41(6):745–752.Article Pubmed
[39]Rahman KM, Aranha O, Sarkar FH. Indole-3-carbinol (I3C) induces apoptosis in tumorigenic but not in nontumorigenic breast epithelial cells. Nutr Cancer. 2003; 45(1):101–112.Article Pubmed
[40]He YH, Friesen MD, Ruch RJ, Schut HA. Indole-3-carbinol as a chemopreventive agent in 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP) carcinogenesis: inhibition of PhIP-DNA adduct formation, acceleration of PhIP metabolism, and induction of cytochrome P450 in female F344 rats. Food Chem Toxicol. 2000; 38(1):15–23.Article Pubmed
[41]Howells LM, Gallacher-Horley B, Houghton CE, Manson MM, Hudson EA. Indole-3-carbinol inhibits protein kinase B/Akt and induces apoptosis in the human breast tumor cell line MDA MB468 but not in the nontumorigenic HBL100 line. Mol Cancer Ther. 2002; 1(13):1161–1172.Article Pubmed
[42]Rahman KM, Aranha O, Glazyrin A, Chinni SR, Sarkar FH. Translocation of Bax to mitochondria induces apoptotic cell death in indole-3-carbinol (I3C) treated breast cancer cells. Oncogene. 2000; 19(50):5764–5771.Article Pubmed
[43]Kassie F, Matise I, Negia M, Upadhyaya P, Hecht SS. Dose-dependent inhibition of tobacco smoke carcinogen-induced lung tumorigenesis in A/J mice by indole-3-carbinol. Cancer Prev Res (Phila). 2008; 1(7):568–576.Article Pubmed
[44]Veeranki OL, Bhattacharya A, Tang L, Marshall JR, Zhang Y. Cruciferous vegetables, isothiocyanates, and prevention of bladder cancer. Curr Pharmacol Rep. 2015; 1(4):272–282.Article Pubmed
[45]Cover CM, Hsieh SJ, Cram EJ, Hong C, Riby JE, et al. Indole-3-carbinol and tamoxifen cooperate to arrest the cell cycle of MCF-7 human breast cancer cells. Cancer Res. 1999; 59(6):1244–1251.Article Pubmed
[46]Paik WH, Kim HR, Park JK, Song BJ, Lee SH, et al. Chemosensitivity induced by down-regulation of microRNA-21 in gemcitabine-resistant pancreatic cancer cells by indole-3-carbinol. Anticancer Res. 2013; 33(4):1473–1481.Article Pubmed
[47]Lyn-Cook BD, Mohammed SI, Davis C, Word B, Haefele A, et al. Gender differences in gemcitabine (Gemzar) efficacy in cancer cells: Effect of indole-3-carbinol. Anticancer Res. 2010; 30(12):4907–4913.Article Pubmed
[48]Bodmer M, Becker C, Meier C, Jick SS, Meier CR. Use of metformin and the risk of ovarian cancer: A case-control analysis. Gynecol Oncol. 2011; 123(2):200–204.Article Pubmed
[49]Bodmer M, Becker C, Meier C, Jick SS, Meier CR. Use of antidiabetic agents and the risk of pancreatic cancer: A case-control analysis. Am J Gastroenterol. 2012; 107(4):620–626.Article Pubmed
[50]Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005; 330(7503):1304–1305.Article Pubmed
[51]Monami M, Colombi C, Balzi D, Dicembrini I, Giannini S, et al. Metformin and cancer occurrence in insulin-treated type 2 diabetic patients. Diabetes Care. 2001; 34(1):129–131.Article Pubmed
[52]Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes Control. 2009; 20(9):1617–1622.Article Pubmed
[53]Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: time for action. J Clin Oncol. 2009; 27(20):3271–3273.Article Pubmed
[54]Currie CJ, Poole CD, Jenkins-Jones S, Gale EA, Johnson JA, et al. Mortality after incident cancer in people with and without type 2 diabetes: Impact of metformin on survival. Diabetes Care. 2012; 35(2):299–304.Article Pubmed
[55]Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, et al. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010; 33(2):322–326.Article Pubmed
[56]Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009; 52(9):1766–1777.Article Pubmed
[57]Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010; 17(2):351–360.Article Pubmed
[58]Grenader T, Goldberg A, Shavit L. Metformin as an addition to conventional chemotherapy in breast cancer. J Clin Oncol. 2009; 27(35):e259.Article Pubmed
[59]Lee JH, Jeon SM, Hong SP, Cheon JH, Kim TI, et al. Metformin use is associated with a decreased incidence of colorectal adenomas in diabetic patients with previous colorectal cancer. Dig Liver Dis. 2012; 44(12):1042–1047.Article Pubmed
[60]Spillane S, Bennett K, Sharp L, Barron TI. A cohort study of metformin exposure and survival in patients with stage I-III colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2013; 22(8):1364–1373.Article Pubmed
[61]Zhang ZJ, Zheng ZJ, Kan H, Song Y, Cui W, et al. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care. 2011; 34(10):2323–2328.Article Pubmed
[62]Schneider MB, Matsuzaki H, Haorah J, Ulrich A, Standop J, et al. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001; 120(5):1263–1270.Article Pubmed
[63]Braghiroli MI, de Celis Ferrari AC, Pfiffer TE, Alex AK, Nebuloni D, et al. Phase II trial of metformin and paclitaxel for patients with gemcitabine-refractory advanced adenocarcinoma of the pancreas. Ecancermedicalscience. 2015; 9:563.Article Pubmed
[64]Suissa S, Azoulay L. Metformin and the risk of cancer: Time-related biases in observational studies. Diabetes Care. 2012; 35(12):2665–2673.Article Pubmed
[65]Pollak MN. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012; 2(9):778–790.Article Pubmed
[66]He YH, Schut HA. Inhibition of DNA adduct formation of 2-amino-1-methyl-6-phenylimidazol [4,5-b] pyridine and 2-amino-3-methylimidazo [4,5-f] quinolone by dietary indole-3-carbinol in females rats. J Biochem Mol Toxicol. 1999; 13(5):239–247.Article Pubmed
 Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved