



- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Cancer Research & Therapy
Volume 6, Issue 6, November 2018, Pages 41–51
HypothesesOpen Access
The acceleration of metastases after tumor removal and the paradoxical phenomenon of concomitant tumor resistance
-
Daniela R. Montagna1,
Paula Chiarella1,
Roberto P. Meiss2 and
Raúl A. Ruggiero1,*

*Corresponding author: Raul A. Ruggiero, Department of Experimental Oncology, IMEX-CONICET, Academia Nacional de Medicina, Pacheco de Melo 3081 (1425), Buenos Aires, Argentina. Tel.: 54-11-4805-3411; E-mail: ruloruggiero@yahoo.com.ar
Received 17 August 2018 Revised 29 September 2018 Accepted 11 October 2018 Published 23 October 2018
DOI: http://dx.doi.org/10.14312/2052-4994.2018-7
Copyright: © 2018 Montagna DR, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
Although surgical extirpation of tumors is usually clinically recommended, tumor removal may entail an undesired side effect: the risk of accelerating the growth of metastases. This effect may account for the relatively modest survival benefits observed when surgery is accomplished after tumor cells have already disseminated to distant anatomical sites even when tumor removal is combined with chemotherapy or immunotherapy. Although different mechanisms could contribute to the enhancement of residual tumor growth after tumor removal, probably a main effect is associated with the withdrawing of an inhibitory effect generated, by certain circumstances, by the primary tumor on its own metastases. This inhibitory effect is a particular case of a more general and paradoxical phenomenon known as concomitant tumor resistance (CR) in which a tumor-bearing host inhibits or retards the growth of secondary tumor implants despite the fact that the primary tumor grows progressively. In this essay we especially focus on the last investigations of our laboratory concerning the importance of tyrosine isomers as mediators of the phenomenon of CR and on their capacity to inhibit established metastases. Taking into account that metastases are considered the main problem in cancer pathology, our investigations aimed to elucidate the molecular basis of the phenomenon of CR might stimulate the design of new and less harmful means of managing malignant diseases, especially by controlling the growth of metastases after the removal of a primary tumor, or after other injuries or stressors that have been claimed to promote the escape of metastases from dormancy.
Keywords: metastases; tumor removal; concomitant tumor resistance; tyrosine isomers
The critical problem of metastasesTop
Metastatic growth is considered a far more serious problem than the original tumor because, for most cases, they ultimately prove to be fatal for the patient. In effect, prior to metastases, most cancers could be cured surgically and 5-year survival rates are estimated at about 90%. However, when a tumor has spread to different sites, those rates, even using some forms of systemic therapy (for example, chemotherapy or immunotherapy), often fall below 15% [1]. The true problem is that there is no assurance that an apparently localized cancer may be known “prior to metastases”. This means that a patient diagnosed with an apparently localized cancer has no guarantee that their tumor has not already spread. For example, in as many as one-third of patients diagnosed with localized breast cancer, carcinoma cells have already disseminated to distant anatomical sites at the time of initial diagnosis. However, these metastatic cells remain undiscovered by the current technics of detection until they awake and ultimately cause the death of the patients [2]. In other cases, the situation is worse. For example, more than 90% of patients with pancreatic cancer have already detected or undetected metastases at the time of initial diagnosis [3]. The management of cancer has improved since the declaration of “War against cancer” in 1971. This improvement was partly due to early detection of the disease and partly due to better therapies. However, the progress has been, in most cases, much slower than originally expected, mainly associated with the difficulty of treating disseminated cancer [4, 5].
These considerations mean that the elucidation of mechanisms aimed to control the growth of metastases are of pivotal importance to design new and more efficient therapeutic strategies against cancer. In the following section we will consider some experimental and clinical evidence that might help to unveil the nature of some of these mechanisms.
Tumor removal and growth of metastasesTop
Experimental evidence
Accelerated growth of metastases following excision of the primary tumor was described more than a century ago by Tyzzer [6]. He observed that, although the surgical removal of a primary tumor prolonged the survival of mice, the size of the individual metastatic nodules was larger than in tumor-bearing mice. Similar results were obtained a decade later by Tadenuma and Okonogi [7] also working with murine models. In the last 90 years, these pioneering experiments were confirmed in many tumor models in which metastases were either detectable or undetected (occult metastases) at the time of tumor removal [8, 9].
As a whole, this large series of experiments can be summarized as follows:
When subcutaneous tumors were removed before the establishment of metastases, the surgery was curative. On the other hand, when metastases have already spread and settled in different tissues and organs, surgery failed to cure the animals and the outcome of that procedure was dependent on the size of the local tumor at the time of removal. Usually, when small tumors were concerned, the lungs and other organs were left with very few metastatic cells as compared with those in tumor-bearing animals in which the primary tumor continued to shed numerous cells into the circulation. In consequence, the total mass of proliferating metastatic cells in tumor-bearing animals exceeded the growth of the fewer cells existing in tumor-excised animals. At this stage, tumor excision significantly prolonged the survival of the mice. When mid-sized tumors were removed, equilibrium could be reached between the enhanced effect on metastases exerted by the surgical extirpation of the primary tumor and the shedding of new potentially metastatic cells in tumor-bearing mice. In consequence, the total mass of proliferating metastatic cells was similar in both tumor-bearing and tumor-excised animals because although tumor-excised animals displayed fewer metastatic foci, each focus was of larger size. At this stage, tumor removal still, although modestly, prolonged the survival of the operated animals, presumably because even though both metastatic masses were similar, the presence of the primary growing tumor was deleterious for the health of the host. Finally, when large tumors were removed, a higher number and size of visible metastatic nodules than those present in tumor-bearing animals, were observed. This reflects the fact that, at that stage, tumor excision would promote the growth of visible metastatic foci as it would also induce non-visible ones to be large enough to be countable. In contrast, in the intact tumor-bearing mice the growth of visible metastases would be limited and there would be no a growth-promoting stimulus for the emergence of new visible metastases from very small undetectable ones. At this stage, tumor excision resulted in a significantly reduced survival of the operated animals [8–17].
Clinical evidence
In clinical settings it is not easy to evaluate the impact of tumor removal on the kinetics of metastatic growth because surgery is one of the primary treatment modalities for solid tumors. In consequence, studies comparing metastatic growth in patients with non-excised tumors (expectant management) with those after tumor resection (surgical management) are very infrequent, although some of them are available in the literature. As a whole, these studies together with indirect evidence accumulated for the last 40 years, suggest that the accelerated growth of metastases following excision of the primary tumor mentioned in the precedent section (see Experimental evidence) may also be associated with human cancer.
For example, Iversen [18] found no benefit with radical prostatectomy over expectant management for adenocarcinoma of the prostate in a follow-up study which followed 111 patients for 23 years.
Similarly, Demicheli [19, 20]; Retsky [21] and Desmedt [22] examined the death-specific hazard rates in patients with breast cancer that had undergone mastectomy alone with those of non-operated patients obtained from an accepted historical database. The non-operated patients (expectant management) exhibited a single peak between the fourth and the fifth year in the hazard rate for death. In contrast, a two peak hazard was detected in the operated patients: the first occurred between the third and the fourth year associated with metastases emerged rapidly between 6 and 12 months after surgery followed by a second peak at the eighth year associated with metastases that emerged about 5-6 years after surgery. These experiments suggest that the natural history of breast cancer could, in some way, be adversely affected by removal of primary tumor, although its negative side effects are, in general, lower than its beneficial ones. In effect, up to date, the clinical studies showed that in most cases, tumor removal actually prolongs the survival of patients because the percentage of operated patients that died early (between the third and the fourth year after surgery) and late (eighth year) is significantly lower than that of non-operated patients. However, a recent debate concerning the utility of primary tumor removal in patients with breast cancer that present with distant metastases (stage IV) at diagnosis, has highlighted the problem in human cancer [23]. Up to date, the clinical studies directed to solve this controversy showed that tumor removal may improve the survival in patients with breast cancer with stage IV, but only in those displaying small primary tumors and limited metastatic load. When larger primary tumors and more metastatic load are concerned, surgery is not recommended [23].
Similar observations have been made concerning colorectal carcinomas. In effect, reported data from the literature support the view that primary tumor resection (PTR) in colorectal cancer with synchronous unresectable metastases should be discussed and validated by a phase III trial in selected patients exhibiting asymptomatic primary tumor, age ≤ 70 years, World Health Organization performance status (WHO-PS) < 2, no extra-hepatic metastatic disease and liver burden of less than 50%. In these patients, PTR, when performed laparoscopically and after preoperative immuno-nutrition, may lead to an increased overall survival. In all other cases, reported postoperative mortality and morbidity rates related to PTR are high and up-front chemotherapy with the primary tumor left in place may represent the more reasonable option [24]. In another study, a total of 116 patients with synchronous colorectal liver metastases were identified of which 49 received an upfront primary tumor resection and 67 received neo-adjuvant chemotherapy. The conclusion of this study indicated that tumor resection resulted in progressive disease suggesting that metastatic growth was enhanced after tumor removal [25].
In many other cancers, it has not been possible to directly evaluate the kinetics of metastatic growth after primary tumor removal because of the lack of control non-operated patients. However, incidental but suggestive evidence has been reported. For example, Sugarbaker [26] reported a clinical case of a 26 year-old male with a melanoma in the scalp. The disease was clinically localized and evaluation revealed no disseminated metastases. A wide excision and graft was performed; six weeks after the operation, numerous subcutaneous nodules as well as visceral metastases appeared and the patient died shortly after. In the same way, partial spontaneous regression of a primary melanoma is actually considered a bad prognosis sign [12, 27]. Lange [28] reported a study of eight patients who underwent cytoreductive surgery for testicular cancer; in each case, the surgical procedure led to a very faster growth of regional and distant residual disease than that expected by assuming an uninterrupted natural growth of these residual tumors. Similar findings in patients with epithelial ovarian cancer [29] led to some investigators to urge caution with respect to cytoreductive surgery. The above clinical studies, together with similar investigations carried out with patients affected by similar or other malignancies strongly suggest that acceleration of metastatic growth may be the undesired outcome of surgical removal of many common human cancers such as melanomas, osteosarcomas and breast, testicular, ovarian, lung, colorectal and bladder carcinomas [8, 12, 30, 31].
Mechanisms associated with the early acceleration of metastases upon tumor removalTop
As a whole, the above mentioned experimental and clinical results are in agreement with the fact that the removal of a tumor in animals or human beings prolonged, in most cases, their survival, especially when the tumors are not large and metastatic load is not high. An obvious advantage of surgical treatment is the reduction of levels of circulating tumor cells released by the tumor, which can be seeded as metastatic foci. In addition, surgical resection can reduce different symptoms including pain, ulceration and lymphoedema that may adversely impact quality of life and function and can also reduce potential immunosuppressive factors released by the primary tumor that may affect putative anti-tumor immune responses. However, on the other hand, a putative disadvantage of surgery is based on the fact that it can promote the progression of metastases. In consequence, we suggest that coupling surgery with treatments aimed to restrain metastatic growth after surgery might, significantly improve the current survival of cancer patients. Some metastases seem to emerge rapidly or relatively rapid after surgery. Others appear after a long time - some months in mice and many years in humans - after tumor removal. Herein, we will discuss only the putative mechanisms underlying the rapid emergence of metastases after surgical tumor excision because they are supposedly more related to the process of tumor withdrawing. On the other hand, the late emergence of metastases or residual tumors may probably be associated with other explanations that have been accounted by our group and others elsewhere [5, 32–34].
The mechanisms claimed to underlie the rapid emergence of metastases after surgical tumor excision may broadly be divided into two groups: A) The presence of a primary tumor does not exert any inhibitory influence on metastases and enhancement of metastatic growth after tumor removal is an emergent phenomenon associated with the injury related to the withdrawing of the tumor; B) The presence of a primary tumor exerts an inhibitory or growth-retarding effect on metastases and enhancement of metastatic growth after tumor removal is only the consequence of the withdrawing of that inhibitory influence.
A) A relatively simple mechanism through which surgery could trigger early metastatic relapse was suggested more than 20 years ago and may derive from the release of cancer cells from the surgical bed during the process of resection [35].
More recently, it has been claimed that surgery up-regulates adhesion molecules in target organs, recruits immune cells capable of entrapping tumor cells and induces changes in cancer cells themselves to enhance migration and invasion to establish at the target site [36].
In addition, surgical trauma induces local and systemic inflammatory responses that can also contribute to the accelerated growth of residual and micro-metastatic disease [36]. Recently, Krall [2] suggested that the systemic inflammatory response induced after surgery associated with the post-surgical wound-healing response promoted the emergence of metastases whose growth was otherwise restricted by a tumor-specific immune T-cell response. In support of this contention, these authors demonstrated that peri-operative anti-inflammatory treatment significantly reduced tumor outgrowth in their model but only when the experiments were carried out with an artificially-induced strongly immunogenic tumor growing in immune-competent mice but not in immune-incompetent NOD/Scid mice. These results are in agreement with previous clinical studies that demonstrated that some anti-inflammatory analgesics – in particular the non-steroidal anti-inflammatory ketorolac – reduced the incidence of early metastatic recurrence in breast cancer patients [22, 37]. It is worth to note, however, that there are some doubts with this interpretation since treatment with other non-steroidal anti-inflammatory agents – for example diclofenac – did not produce such anti-metastatic effect [22]. In fact, Guo [38] suggested that the anti-metastatic effects of ketorolac could be attributed to its ability to diminish tumor cell adhesion, migration and invasion rather than to its anti-inflammatory properties.
B) Although the mechanisms mentioned in item A) could contribute to the enhancement of residual tumor growth after tumor removal in some experimental and human tumors, they probably do not represent the whole story. In effect, in a number of experiments carried out for many years, control series were included that demonstrated that accelerated growth of metastases following tumor excision was not due or was not mainly due to high release of tumor cells, stress, surgical trauma or anesthesia. This conclusion is supported by three kinds of experiments: First, sham operations including surgical procedures carried out in the local area without removing the tumors, or amputation of a non-tumor bearing leg leaving intact the tumor in the other leg, had no influence on the metastatic growth [9, 11, 17, 39–41]. Second, accelerated metastatic growth after tumor extirpation can be abrogated by re-inoculation of cells of the removed tumor. This abrogation was achieved when high doses of tumor cells were re-inoculated and local tumor reached the size at which the inhibitory effect putatively exerted or induced by the tumor was fully expressed. On the other hand, re-inoculation of small doses of tumor cells had no influence on the growth of metastases, presumably because the latent period for tumors was too long to influence the growth of metastatic cells already settled in lung or elsewhere [9, 13, 42]. Third, experimental secondary tumor implants carried out by the subcutaneous, intravenous or intra-peritoneal routes were strikingly inhibited by the presence of a primary tumor in both immune-competent and immune-deficient mice. In these experiments, either immunogenic or non-immunogenic murine tumors, as well as human tumors growing in immune-deficient mice were used. These experiments suggested that a primary tumor can restrain the growth of metastases that can be considered as natural secondary tumor implants developed spontaneously during the growth of a primary tumor [9, 15, 16, 42–49]. These experiments also demonstrated that the mechanisms of enhancement of metastases after tumor removal would not be necessarily dependent upon a previous immune restriction of the metastatic foci because the inhibition produced by the primary tumor on its metastases can be immunological, as well as non-immunological in nature.
The phenomenon of concomitant tumor resistance
Taken together, the available evidence suggests that, although other mechanisms may affect the fate of metastases after the removal of a primary tumor, probably a main effect is associated with the withdrawing of an inhibitory effect generated, upon certain circumstances, by the primary tumor on its own metastases. This inhibitory effect is a particular case of a more general phenomenon known as concomitant tumor resistance. Concomitant tumor resistance or, more briefly, concomitant resistance (CR) is the paradoxical phenomenon according to which a tumor-bearing host inhibits or retards the growth of secondary tumor implants despite the fact that the primary tumor grows progressively [12, 50]. CR was originally described by Paul Ehrlich at the turn of the 20th century [51] but, apart from a few isolated reports [52, 53] it remained virtually forgotten for about 60 years until it was re-discovered in the 60’s by Southam [54], Gershon [55] and others [56, 57]. Since that moment on, some groups have studied this phenomenon demonstrating that both immunogenic and non-immunogenic tumors can induce CR in different animal models such as mice, rats and hamsters [9, 16, 17, 45, 58, 59]. However, even after its renascence, CR has not received much attention as compared with other areas of cancer research despite the fact that it has been directly described in human beings [54] and, as shown above, despite its putative relevance to the mechanisms of metastases control. At first sight, this lack of attention in the phenomenon of CR may be related to the fact that its study implies the search for anti-tumor mechanisms that depend on the presence of a primary growing tumor. This apparently paradoxical approach does not seem to have been attractive and promissory for many.
Although the phenomenon of CR against the growth of metastases has been observed in many experimental systems and it has also been suspected in many clinical situations, on the other hand, there are also some experimental and clinical evidence that the presence of a tumor may not exert any effect or even a stimulating effect (Concomitant enhancement, CE) on their metastases [60–64]. In such cases, tumor removal would not induce any acceleration or even it would induce an inhibition of metastatic growth. In clinical settings, few putative examples of CE have been reported. Most of them have been related to occasional suspected regressions of hepatic and/or pulmonary metastases following nephrectomy for renal cell carcinoma [65–68]. In our laboratory, we have demonstrated the presence of both CR and CE phenomena in some tumor-bearing mice, depending on the ratio between the mass of the larger primary tumor mass relative to that of the smaller secondary one, with high ratios rendering inhibition and low ratios inducing stimulation of the secondary tumor. However, in our experience, the magnitude of this stimulatory effect, whenever it is present, proved to be rather modest as compared with the magnitude of the inhibitory effect [69]. A similar conclusion may be attained by analyzing the data from other laboratories [47, 64].
In consequence, taken together, the available experimental and clinical evidence suggest to us that CR would be more likely than CE to govern the behavior of animal as well as commonly occurring human tumors.
Mechanisms associated with the phenomenon of concomitant tumor resistanceTop
Historical background
Different hypothesis have been proposed to explain the phenomenon of CR
According to the immunological hypothesis, the growth of a tumor generates a specific antitumor immune response which even though it is not strong enough to inhibit the primary tumor growth, is still capable of preventing the development of a relatively small secondary tumor inoculum. This explanation is not very different from that of conventional immunological rejection of allogeneic tumors in naive mice or immunogenic syngeneic tumors in pre-immunized animals. The immunological hypothesis was originally proposed by Bashford [52] which, in turn, coined the term “concomitant immunity” by which this phenomenon has been known in the past.
This interpretation is supported by solid evidence mainly based on experiments with strongly immunogenic murine tumors induced by chemical agents or viruses [59, 70]. However, it does not provide a satisfactory explanation for the fact that CR has also been observed in association with spontaneous murine tumors of non-detectable immunogenicity [16, 17, 58, 59].
Non-immunological explanations rely mainly on two hypotheses
Ehrlich [51] and Tyzzer [6] believed that nutrients essential for tumor growth are consumed by the primary tumor, making it difficult or impossible for a second implant to develop (atrepsis theory). The term “atrepsis” was coined by Joseph Parrot in 1874 to describe malnutrition, especially in infants [71]. A support for the atrepsis theory to explain CR is associated with the fact that a progressive tumor is a trap for glucose, nitrogen and other nutrients. In this way, all attempts to correct the weight loss in tumor-bearing organisms by supplying different nutrients by the intravenous route resulted in acceleration of tumor growth [9]. Taking into account that there is convincing evidence that restriction of nutrients may be accompanied by inhibition of tumor growth, it is possible that in the setting of a severe systemic biochemical disturbance generated by the primary tumor, the condition for the proliferation of re-inoculated tumor cells (secondary tumor implants) cannot be as favorable as in control animals.
Others [9, 48, 72–74] have postulated that the primary tumor produces or induces the production of anti-proliferative nonspecific substances or anti-angiogenic molecules which limit the replication of tumor cells of the second inoculum. The idea that a tumor induces systemic effects by the production of some kind of substances was originally suggested by Nakahara and Fukuoka in the 50’s in their concept of cancer toxohormone, whose circulating concentration should rise with increased tumor mass [75].
Taken together, these non-immunological hypotheses can offer a putative explanation for the CR induced by non-immunogenic tumors but not for the specific inhibition of secondary tumor implants observed during the growth of immunogenic tumors.
The two main peaks of CR throughout tumor growth
For the last 30 years, we have studied, in our laboratory, the phenomenon of CR associated with the growth of many murine and human tumors (the latter growing into immune-deficient mice) in an attempt to integrate the different hypotheses into a coherent picture. Our results, summarized in Table 1 and reported, at least in part, in former papers [17, 45, 49, 59, 73, 76, 77] demonstrated that, two main temporally separate peaks or events of CR are generated during primary tumor growth. The first peak was only induced by immunogenic tumors of small size (≤ 500 mm3); it was tumor-specific and thymus-dependent as it was exhibited in euthymic but not in nude mice, its intensity was proportional to tumor immunogenicity and a typical immunological rejection - associated with extensive necrosis and a profuse infiltration with polymorphonuclear granulocytes and mononuclear cells was observed histologically at the site of the second tumor implant undergoing CR. Furthermore, the kinetics of appearance and disappearance of the first peak of CR paralleled the kinetics of appearance and disappearance of specific cytotoxic antibodies and cell-mediated cytotoxicity against the tumors.
| Tumor | Origin | Immunogenicity | Concomitant tumor resistance | |
| 1o Peak | 2a Peak | |||
| Murine | ||||
| L15-A1 | Allogeneic | Very strong | Very high | Very high |
| MC-D2 | Induced by MCa | Very strong | Very high | Moderate |
| MC-C2 | Induced by MCa | Very strong | High | High |
| MNU-MPA3 | Induced by MNU-MPAb | Moderate | Moderate | Moderate |
| MC-B2 | Induced by MCa | Moderate | Moderate | Moderate |
| S-180-O2 | Spontaneous | Moderate | Moderate | Moderate |
| MNU3 | Induced by MNUc | Weak | Low | Moderate |
| M33 | Spontaneous | Weak | Low | Moderate |
| LMM33 | Spontaneous | Weak | Low | Absent |
| CS3 | Induced by MMTVd | Weak | Low | High |
| C7HI3 | Induced by MPAe | Undetectable | Absent | Absent |
| PX2 | Induced by GCf | Undetectable | Absent | Moderate |
| S-180-N2 | Spontaneous | Undetectable | Absent | Moderate |
| P3881 | Induced by MCa | Undetectable | Absent | Very high |
| CM3 | Spontaneous | Undetectable | Absent | High |
| CEP3 | Spontaneous | Undetectable | Absent | High |
| CEI3 | Spontaneous | Undetectable | Absent | High |
| CPV3 | Spontaneous | Undetectable | Absent | Moderate |
| L15-S1 | Spontaneous | Undetectable | Absent | High |
| LB1 | Spontaneous | Undetectable | Absent | Very high |
| Human | ||||
| KBα | Spontaneous | ----------- | Absent | High |
| Calu-6β | Spontaneous | ----------- | Absent | High |
| PC3γ | Spontaneous | ----------- | Absent | Very high |
Abbreviations: 1Lymphoma-leukemia; 2Fibrosarcoma; 3Carcinoma; aMC = Methylcholanthrene; bMNU-MPA = N-methyl-N-nitrosourea + medroxyprogesterone acetate; cMNU = N-methyl-N-nitrosourea; dMMTV = Murine mammary tumor virus; eMPA = medroxyprogesterone acetate; fGC = glass cylinder; αNasopharyngeal carcinoma; βLung carcinoma; γProstatic carcinoma. More details of the tumors are given elsewhere: see quotations [8, 12, 13, 15-17, 45, 49, 59, 76, 77].
On the other hand, the second peak of CR was induced by both immunogenic and non-immunogenic large tumors (≥ 2000 mm3); it was tumor-non-specific and thymus-independent as it was exhibited in both euthymic and nude mice, it did not correlate with tumor immunogenicity and its intensity was proportional to the primary tumor mass: the larger the primary tumor, the stronger the inhibition of the secondary tumor. Further, the inhibition of the secondary tumor by a large primary tumor was neither associated with a massive or focal necrosis nor with any host cell infiltration, contrasting with a classical immunological rejection. Instead, the secondary tumor implant remained in a dormant-like state, with viable but non-infiltrating tumor cells placed at the inoculation site between the skin and the muscular layer. Occasionally some apoptotic tumor cells began to appear after 24 h of inhibition.
Some years ago, an intermediate peak of CR was reported to be associated with a particular type of mid-sized tumors (1,000 - 1,500 mm3) – typically the Lewis lung carcinoma that restrain secondary tumors indirectly, by limiting tumor neovascularization [72]. Although the mechanisms associated with the first and intermediate peaks of CR have been elucidated as T cell–dependent and angiostatin-dependent, respectively, the molecular basis of the most universal manifestation of CR, that is, the second peak, remained an enigma for many years.
In former studies, we demonstrated that the intensity of the second peak of CR correlated with the activity of a serum factor (or factors), different from antibodies, complement or other well characterized growth inhibitory molecules, that inhibited the in vitro and in vivo proliferation of tumor cells. Further, mice bearing tumors that produced CR and such inhibitory serum factors, could have or not have metastases, but in the case of having detectable or undetectable metastases, metastatic growth was strongly enhanced after tumor removal. Reciprocally, when the serum inhibitory activity was absent – the only two cases were mice bearing two highly metastatic mammary carcinomas – the second peak of CR did not appear [13, 15, 59]. Further, after surgical extirpation of these tumors, growth of metastases was not stimulated.
These results suggested a direct correlation among the second peak of CR, the capacity to restrain the growth of metastases and the titer of serum growth inhibitory activity. Very interestingly, metastases produced by the two tumors that did not produce CR, were significantly inhibited by both the concomitant presence of unrelated tumors that induced CR and by the daily administration of serum from mice bearing these unrelated tumors, which displayed a high titer of growth inhibitory activity.
In recently published works [17, 45], we identified the anti-tumor serum factors associated with CR as a rather equi-molar mixture of meta-tyrosine (m-tyr) and ortho-tyrosine (o-tyr), two unnatural isomers of tyrosine, unnatural meaning that it is thought that they are absent from normal proteins [78, 79]. We carried out this characterization starting from mice bearing a non-immunogenic murine lymphoma that produces the strongest second peak of CR among all our murine tumor models [17] and also from nude mice bearing a human prostatic carcinoma that produces the strongest CR among all the human lines tested [45]. We could demonstrate that m-tyr and o-tyr were responsible for 90% and 10%, of the total antitumor activity of the serum, respectively, as determined by the inhibition of both the in vitro proliferation of different murine and human tumor cells and the in vivo growth of subcutaneous tumor implants.
Therapeutic effect of m-tyr and o-tyr on established metastases of murine and human originTop
The first evidence of the therapeutic value of m-tyr and o-tyr on established metastases was obtained using mice bearing two different murine metastatic mammary tumors growing subcutaneously. These tumors did not produce CR against experimental secondary tumor implants but they were very sensitive to the CR induced by unrelated tumors. When a periodic treatment of these tumor-bearing mice with m-tyr or o-tyr was initiated at the time when metastatic foci were already present in both lung and liver as demonstrated in control sacrificed tumor-bearing mice - a striking inhibition of metastatic growth was observed as evaluated in tumor-bearing mice that were sacrificed three and four weeks after the onset of the treatment [45, 49, 80, 81].
Similar results were observed on human metastatic cells growing in immune deficient mice [45, 49]. In one series of experiments, the inhibitory effect of m-tyr was achieved against experimental metastases produced in the lung by the intravenous inoculation of cells from a human prostatic tumor line; the rationale to use experimental metastases was due to the fact that this human tumor line growing subcutaneously does not produce metastases spontaneously neither in nude nor in NOD/Scid gamma mice. In another series of experiment, using a human breast carcinoma that produces metastases spontaneously in NOD/Scid gamma mice, periodic treatment with m-tyr resulted in a significant inhibition of spontaneous lung metastases as evaluated in mice that were sacrificed 3 weeks after the onset of the treatment.
The most impressive evidence of the therapeutic value of m-tyr and o-tyr was obtained in an experiment aimed to mimic a putative clinical situation. A highly metastatic mammary murine tumor - called LMM3 - was surgically removed at the time when metastases had already settled in lung and liver. Then, a daily inoculation of either m-tyr or o-tyr by the intravenous route was carried out for 35 consecutive days starting one day after surgery. The results revealed a striking anti-metastatic effect produced by both tyrosine isomers although the effect produced by m-tyr was significantly more robust than that of o-tyr. In effect, all controls (9 out of 9) died rapidly (median of survival = 29 days after surgery), exhibiting high number of lung and hepatic metastases. In contrast, only two m-tyr treated-mice died (at days 29 and 97 after surgery), while the other 6 mice remained alive without exhibiting signs of local or metastatic disease for the rest of their lives: when these mice were sacrificed at 22 months old (that is about 18 months after the end of the treatment) no metastatic foci were detected neither in lung nor in liver nor elsewhere. As for o-tyr treated mice, all mice (5 out of 5) died although significantly later (median of survival = 80 days after surgery) than controls [49, 80]. For illustrative purposes of this experiment, we show the Figure 1 that has already been published by our group in Strazza [49].
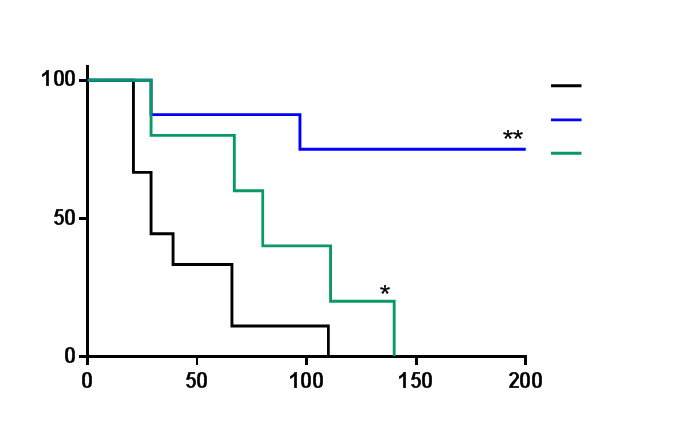
When treatment with m-tyr was initiated later, when the metastatic load were three times larger than in the first experiment, a significant anti-metastatic effect was also achieved and a significant percentage of treated mice (50%) survived at least six months after all controls had died [45].
The therapeutic value of these and similar experiments is stressed by the fact that the striking antitumor effects mediated by m-tyr and o-tyr were observed without exhibiting any detectable toxic-side effects even using doses 20 times higher than therapeutic ones [45, 49, 80].
Origin of tyrosine isomers and putative mechanisms of tumor inhibitionTop
To date, m-tyr and o-tyr have been studied almost exclusively as markers for oxidative damage associated with abnormal proteins as detected in, for example, the blood of animals subjected to cardiac ischemia-reperfusion injury, mitochondria of exercised animals, atherosclerotic tissue of diabetic primates, and aging lens of humans [79]. Most investigators have assumed that m-tyr and o-tyr are generated post-translationally when the L-phenylalanine present in proteins is exposed to hydroxyl radicals during oxidative damage. However, it was recently suggested that oxidized amino acids, such as m-tyr and o-tyr, may also be generated from free amino acids that subsequently could be incorporated into proteins during synthesis [78, 79].
In previous experiments [17], we observed that the serum antitumor activity attributed to m-tyr and o-tyr was strongly inhibited by agents that reduced the number of myeloid-derived suppressor cells (MDSC) and oxidative damage, and that in many tumor-bearing mice and some cancer patients, MDSCs that produced large amounts of reactive oxygen species accumulated progressively in circulation [17, 82–85]. On the basis of these results, we suggested that free m-tyr and o-tyr present in the serum from tumor-bearing mice will be produced, at least in part, when circulating molecules of phenylalanine are oxidized by hydroxyl radicals released by MDSC.
The inhibition exerted by m-tyr and o-tyr on tumor growth mimics the inhibition produced by CR. In both cases, tumor inhibition was primarily associated with the presence of a high proportion of tumor cells in G0, a decrease in G2-M phases, and an increase or accumulation of cells in the S-phase, considered the consequence of an S phase arrest [17]. Further, the inhibitory effect produced in vitro and in vivo by m-tyr and o-tyr on tumor cell proliferation was counteracted by phenylalanine and, to a lesser degree, by glutamic acid, aspartic acid, glutamine and histidine, but not by tyrosine or the remaining protein amino acids.
A molecular analysis [17, 45] showed that the antitumor effects mediated by m-tyr and o-tyr on murine and human tumor cells were mediated, at least in part, by an early inactivation of p-STAT3 and down regulation of the NFκB//NOTCH axis that are constitutively activated in many tumor cells. Inactivation of STAT3 impaired its nuclear translocation and down regulated the expression of survivin as well as other genes engaged with cell proliferation and survival that are targets of STAT3, such as BCL-XL (B-cell lymphoma XL), cyclin D1 and myc, among others. Taken together, all of these effects could drive tumor cells into a state of dormancy in G0 phase as determined by the low expression of Ki167 protein in tumor cells treated with tyrosine isomers. On the other hand, the S-phase arrest might be generated by a different mechanism that up to date, remains speculative. Several factors and conditions such as resveratrol, hyperoxia, hydroxyurea, ultraviolet radiation, G-rich oligonucleotides and zidovudine, induce the inhibition of cell proliferation associated with an S-phase arrest, presumably by the activation of an intra–S-phase checkpoint [86]. Different mechanisms for activating this checkpoint have been proposed, including accumulation of cdk2 (cyclin-dependent kinase 2) in its inactive phosphorylated form, downregulation of cdk2, activation of ATM/ATR (ataxia telangiectasia mutated/ataxia telangiectasia Rad 3-related) kinase in response to DNA damage, modulation or inhibition of a replicative helicase activity, and downregulation of cyclin A2.
After these primary effects, apoptosis and autophagy were observed in some of the previously arrested tumor cells. Putative mechanisms of activation of autophagy by m-tyr have been studied in our last paper [45]. Previous reports have shown that STAT3 inhibition, such as produced by m-tyr, induces signs of autophagy [87]. Alternatively, m-tyr may be incorporated into eukaryotic proteins via a specific tRNA-dependent pathway, using mitochondrial and possibly cytosolic phenylalanyl-tRNA synthetase [88]. In turn, elevated m-tyr content in proteins may lead to the dysfunction of intracellular signaling such as the inhibition of LC3-II degradation [89] observed in our experiments, which promotes the activation of autophagy [45].
The central paradox of concomitant tumor resistanceTop
The central paradox of CR, i.e., the inhibition of secondary tumor implants together with the progressive growth of the primary tumor, has remained unresolved for more than a century. To account for this problem, we showed that as a primary tumor grew subcutaneously, relatively large amounts of most amino acids, including those that counteract the inhibitory effects of m-tyr and o-tyr (i.e., phenylalanine, glutamic acid, aspartic acid, glutamine and histidine), accumulated in the tumor microenvironment, whereas at a contralateral flank receiving a secondary tumor implant, the content of amino acids was significantly lower. Furthermore, a cocktail of amino acids similar to that detected close to the primary site proved to be more counteracting of the antitumor effects produced by m-tyr and o-tyr than a cocktail similar to that detected at the secondary site [17]. On this basis, we suggested that a secondary tumor can be inhibited by circulating m-tyr and o-tyr, while at the same time, the primary tumor is protected from their inhibitory effects, at least in part, by these counteracting amino acids and thus can continue to grow [17]. This suggestion seems to reconcile the two major non-immunological interpretations of CR that have been advanced to date, i.e., the anti-proliferative-factors hypothesis and the atrepsis theory [9, 51, 72–74]. In effect, the postulation that serum m-tyr and o-tyr are responsible for the inhibitory effect generated by a primary tumor on the growth of secondary tumor implants is similar to the hypothesis of anti-proliferative factors. However, the mere presence of inhibitory factors such as m-tyr and o-tyr is not enough to explain why the primary tumor can grow while the secondary one cannot. On the other hand, the different concentration of amino acids at the site of the primary tumor compared with that at the site of a secondary tumor implant would appear to support the atrepsis theory because, according to this theory, the primary tumor accumulates elements that allow it to grow and whose lack at distant sites from the primary tumor will prevent a second tumor from growing. However, whereas according to the atrepsis theory, these elements are nutrients that would directly stimulate growth of the primary tumor, in our postulation, these elements would allow the primary tumor to grow by counteracting the effect of circulating inhibitory factors. Some years ago, Prehn [50] anticipated this interpretation and suggested that CR could best be explained by the competitive interaction of two opposing - and up to that time uncharacterized - influences: a local, slowly diffusible, tumor-facilitating environment that would be counteracted by circulating inhibitors.
This interpretation might also explain two intriguing questions associated with the biological effects of m-tyr and o-tyr. Data from our and other laboratories suggest that both tyrosine isomers can inhibit not only the in vitro proliferation of tumor cells but also, although probably to a lesser extent, that of normal cells [17, 78, 79]. However, a) the regeneration of normal tissues is usually not affected in tumor-bearing mice that exhibit CR [48, 90], and b) periodic inoculation of m-tyr and o-tyr, that produces a strong inhibition on the growth of metastatic foci, at the same time it does not produce any inhibitory effect neither on normal regenerative processes, development of normal immune responses nor on proliferation in vivo of MDSC that, at least in part, would produce m-tyr and o-tyr in tumor-bearing mice [45, 49]. We could explain these questions by assuming that these normal proliferating tissues, but not secondary tumor implants, display a content of amino acids high enough to counteract the inhibitory effects produced by m-tyr and o-tyr. Experiments underway in our laboratory are aimed to confirm these assumptions.
New avenues for cancer understanding and treatmentTop
Surgical extirpation is the mainstay treatment of solid tumors and may be curative when metastatic cells have not already disseminated from the primary tumor [1]. However, although it is recommended in most clinical cases, tumor removal may entail an undesired side effect i.e., the acceleration of regional and distant (metastases) residual neoplastic disease [36]. This effect may account for the disappointingly modest survival benefits observed when surgery is accomplished when tumor cells have already disseminated to distant anatomical sites even when tumor removal is combined with systemic treatments such as chemotherapy. Investigators have proposed some therapeutic options to limit metastatic growth after tumor removal, including the use of perioperative (instead of postoperative) chemotherapy, antioxidant agents, immunotherapy, and bio-modulation [91], but to date, the results have not been as promissory as expected.
The elucidation of the phenomenon of CR could contribute to overcome this problem on the basis that the mechanisms underlying CR can be considered similar or identical to those utilized for a primary tumor to limit the growth of its own natural secondary tumor implants generically known as ‘metastases’.
However, in the past, CR has usually been rather neglected by researchers and clinicians. As we stated above (see the section called “The phenomenon of concomitant tumor resistance”) this attitude could, at first sight, be due to the fact that the study of CR does not seem to be attractive since it implies to search for mechanisms of tumor inhibition that depend on the presence of a primary growing tumor. Probably, the root of this attitude is more profound. In effect, the idea that a primary tumor may exert inhibitory influences upon distant metastases meant that a tumor had to be considered an integrated, organ-like entity rather than a collection of independent atypical cells as it has been usually considered.
However, there are numerous observations in the literature that support that idea [5, 19, 32, 50, 73, 92–95]. For example, hepatectomy stimulates mitosis in previously resting hepatocytes that had been implanted ectopically, or nephrectomy stimulates the proliferation (and also the hypertrophy) of the contralateral kidney in the same way that excision of a primary tumor induces mitosis in previously arrested secondary tumor implants. Furthermore, different from bacteria and other unicellular organisms which grow exponentially, if nutrients are available, growth of both normal organs and tumors follow a Gompertzian-like curve that is exponential at first and then it is modified by an exponential decline in rate when they approach to an asymptote. This decline proved not to be caused by failure of blood and nutrients supply or any other artifact of increased size. The only difference between a normal organ and a tumor, apart from the tendency of a tumor to metastasize, seems to be that the plateau size of the normal organ is reached when the organ reaches its full size, while the putative plateau size of the tumor would be larger than is compatible with the life of the host [96]. In addition, it has been demonstrated in different murine tumors, that mixtures of particular sub-clones tended, in the resulting tumors, to approach reproducible proportions characteristic for that array of sub-clones and that these final proportions were independent of the starting proportions and of the selective pressures favoring each particular sub-clone [96]. This could hardly have been possible if each particular sub-clone were not in some type of communication with the other sub-clones in order to maintain them in a constant proportion despite different selective pressures.
In a relatively recent paper [17] we have elucidated the serum factors responsible for the most universal manifestation of CR, as a mixture of m-tyr and o-tyr, two unnatural isomers of tyrosine that exhibited strong antitumor effects. In subsequent communications [45, 49, 80, 81], we could demonstrate that both m-tyr and o-tyr could inhibit, in both in vitro and in vivo settings, not only the proliferation of tumor cells derived from tumors that do induce CR, but also that of those derived from tumors that do not induce CR, thus widely increasing their therapeutic possibilities.
The most anti-tumor impressive effect of both tyrosine isomers was achieved on the growth of established metastases of both murine and human origins. Most importantly, these anti-metastatic effects were achieved even at very low concentrations and, different from conventional chemotherapy that usually impairs the health of the body, both m-tyr and o-tyr seemed to exert their anti-tumor effects without displaying any detectable toxic-side effects even using doses 20 times higher than therapeutic ones.
However, more experiments measuring different physiologic variables not only in mice, but also in other species such as rats and rabbits, in acute, sub-acute and chronic schedules of m-tyr and o-tyr administration, will be necessary to demonstrate more accurately their lack of toxic-side effects.
Taken together, all the experiments reported in previous communications, as well as new experiments, that are underway, aimed to explore further the molecular basis of the inhibitory effects of m-tyr and o-tyr on tumor cell proliferation, could help to develop new and less harmful means of managing malignant diseases, especially by controlling the growth of metastases after the removal of a primary tumor, or after other surgical injuries or stressors that have been claimed to promote the escape of metastases from dormancy. This is an objective not yet achieved by current oncology research.
Conflicts of interest
No potential conflicts of interest were disclosed.
ReferencesTop
[1]Chen J, Sprouffske K, Huang Q, Maley CC. Solving the puzzle of metastasis: The evolution of cell migration in neoplasms. PLoS One. 2011; 6(4):e17933.Article Pubmed
[2]Krall JA, Reinhardt F, Mercury OA, Pattabiraman DR, Brooks MW, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Transl Med. 2018; 10(436). pii: eaan3464.Article Pubmed
[3]Freelove R, Walling AD. Pancreatic cancer: Diagnosis and management. Am Fam Physician. 2006; 73(3):485–492.Article Pubmed
[4]Tiwari AK, Roy H. Progress against cancer (1971-2011): How far have we come? J Intern Med. 2012; 27(4):392–399.Article Pubmed
[5]Sonnenschein C, Soto AM. Cancer metastases: So close and so far. J Natl Cancer Inst. 2015; 107(11). pii:djv236.Article Pubmed
[6]Tyzzer EE. Factors in the production and growth of tumor metastases. J Med Res. 1913; 28(2):309–332.Pubmed
[7]Tadenuma K, Okonogi S. Experimentelle Untersuchugen Metastase bei Mausecarcinom. Z Krebsforsch. 1924; 21:168–172.
[8]Ruggiero RA, Bruzzo J, Chiarella P, Bustuoabad OD, Meiss RP, et al. Concomitant tumor resistance: The role of tyrosine isomers in the mechanisms of metastases control. Cancer Res. 2012; 72(5):1043–1050.Article Pubmed
[9]Gorelik E. Concomitant tumor immunity and the resistance to a second tumor challenge. Adv Cancer Res. 1983; 39:71–120.Article Pubmed
[10]Keller R. Repression of lymphatic metastasis by a second implant of the same tumor. Invasion Metastasis. 1985; 5(5):295–308.Article Pubmed
[11]Ketcham AS, Kinsey DL, Wexler H, Mantel N. The development of spontaneous metastases after the removal of a “primary” tumor. Cancer. 1961; 14:875–882.Article Pubmed
[12]Chiarella P, Bruzzo J, Meiss RP, Ruggiero RA. Concomitant tumor resistance. Cancer Lett. 2012; 324(2): 133–141.Article Pubmed
[13]Di Gianni P, Franco M, Meiss RP, Vanzulli S, Piazzon I, et al. Inhibition of metastases by a serum factor associated to concomitant resistance induced by unrelated murine tumors. Oncol Rep. 1999; 6(5):1073–1084.Article Pubmed
[14]Gorelik E. Antimetastatic concomitant immunity. In: Tumor Invasion and Metastasis. Liotta LA and Hart IR, The Hague. Martinus Nijhoff Publishers. 1982; 113–131.
[15]Bonfil RD, Ruggiero RA, Bustuoabad OD, Meiss RP, Pasqualini CD. Role of concomitant resistance in the development of murine lung metastases. Int J Cancer. 1988; 41:415–422.Article Pubmed
[16]Ruggiero RA, Bustuoabad OD, Bonfil RD, Meiss RP, Pasqualini CD. “Concomitant immunity” in murine tumours of non-detectable immunogenicity. Br J Cancer. 1985; 51(1):37–48.Article Pubmed
[17]Ruggiero RA, Bruzzo J, Chiarella P, Di Gianni P, Isturiz MA, et al. Tyrosine isomers mediate the classical phenomenon of concomitant tumor resistance. Cancer Res. 2011; 71(22):7113–7124.Article Pubmed
[18]Iversen P, Madsen PO, Corle DK. Radical prostatectomy versus expectant treatment for early carcinoma of the prostate. Twenty-three year follow-up of a prospective randomized study. Scand J Urol Nephrol Suppl. 1995; 172:65–72.Article Pubmed
[19]Demicheli R, Miceli R, Moliterni A, Zambetti M, Hrushesky WJ, et al. Breast cancer recurrence dynamics following adjuvant CMF is consistent with tumor dormancy and mastectomy-driven acceleration of the metastatic process. Ann Oncol. 2005; 16(9):1449–1457.Article Pubmed
[20]Demicheli R, Retsky MW, Hrushesky WJ, Baum M, Gukas ID. The effects of surgery on tumor growth: A century of investigations. Ann Oncol. 2008; 19(11):1821–1828.Article Pubmed
[21]Retsky MW, Demicheli R, Hrushesky WJ, Baum M, Gukas ID. Dormancy and surgery-driven escape from dormancy help explain some clinical features of breast cancer. APMIS. 2008; 116(7-8):730–741.Article Pubmed
[22]Desmedt C, Demicheli R, Fornili M, Bachir I, Duca M, et al. Potential benefit of intra-operative administration of ketorolac on breast cancer recurrence according to the patient’s body mass index. J Natl Cancer Inst. 2018; 110(10).Article Pubmed
[23]Nguyen DH, Truong PT. A debate on locoregional treatment of the primary tumor in patients presenting with stage IV breast cancer. Expert Rev Anticancer Ther. 2011; 11(12):1913–1922.Article Pubmed
[24]de Mestier L, Manceau G, Neuzillet C, Bachet JB, Spano JP, et al. Primary tumor resection in colorectal cancer with unresectable synchronous metastases: A review. World J Gastrointest Oncol. 2014; 6(6):156–169.Article Pubmed
[25]Slesser AA, Khan F, Chau I, Khan AZ, Mudan S, et al. The effect of a primary tumour resection on the progression of synchronous colorectal liver metastases: An exploratory study. Eur J Surg Oncol. 2015; 41(4):484–492.Article Pubmed
[26]Sugarbaker EV, Thornthwaite J KA. Inhibitory effect of a primary tumor on metastasis. In: Cancer Invasion and Metastasis Biological Mechanisms and Therapy. 1977. p. 227–40.
[27]Prehn RT. The paradoxical association of regression with a poor prognosis in melanoma contrasted with a good prognosis in keratoacanthoma. Cancer Res. 1996; 56(5):937–940.Article Pubmed
[28]Lange PH, Hekmat K, Bosl G, Kennedy BJ, Fraley EE. Accelerated growth of testicular cancer after cytoreductive surgery. Cancer. 1980; 45(6):1498–1506.Article Pubmed
[29]Hoskins WJ. The influence of cytoreductive surgery on progression – free interval and survival in epithelial ovarian cancer. Baillieres Clin Obstet Gynaecol. 1989; 3(1):59–71.Article Pubmed
[30]Kaya M, Wada T, Nagoya S, Kawaguchi S, Isu K, et al. Concomitant tumour resistance in patients with osteosarcoma. A clue to a new therapeutic strategy. J Bone Joint Surg Br. 2004; 86(1):143–147.Article Pubmed
[31]Peeters CF, De Waal RM, Wobbes T, Ruers T. Metastatic dormancy imposed by the primary tumor: Does it exist in humans? Ann Surg Oncol. 2008; 15(11):3308–3315.Article Pubmed
[32]Ruggiero RA, Bustuoabad OD. The biological sense of cancer: A hypothesis. Theor Biol Med Mod. 2006; 3:43.Article Pubmed
[33]Rubin H. What keeps cells in tissues behaving normally in the face of myriad mutations? Bioessays. 2006; 28(5):515–524.Article Pubmed
[34]Linde N, Fluegen G, Aguirre-Ghiso JA. The relationship between dormant cancer cells and their microenvironment. Adv Cancer Res. 2016; 132:45–71.Article Pubmed
[35]Eschwège P, Blanchet P, Benoit G, Jardin A, Dumas F, et al. Haematogenous dissemination of prostatic epithelial cells during radical prostatectomy. Lancet. 1995; 346(8989):1528–1530.Article Pubmed
[36]Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016; 76(6):1367–1380.Article Pubmed
[37]Retsky M, Demicheli R, Hrushesky WJ, Forget P, De Kock M, et al. Reduction of breast cancer relapses with perioperative non-steroidal anti-inflammatory drugs: New findings and a review. Curr Med Chem. 2013; 20(33):4163–4176.Article Pubmed
[38]Guo Y, Kenney SR, Muller CY, Adams S, Rutledge T, et al. R-Ketorolac targets Cdc42 and Rac1 and alters ovarian cancer cell behaviors critical for invasion and metastasis. Mol Cancer Ther. 2015; 14(10):2215–2227.Article Pubmed
[39]Schatten WE, Kramer WM. An experimental study of perioperative tumor metastases. II. Effects of anesthesia, operation and cortisone administration on growth of pulmonary metastases. Cancer. 1958; 11(3):460–462.Article Pubmed
[40]Schatten WE. An experimental study of perioperative tumor metastases. I. Growth of pulmonary metastases following total removal of pulmonary leg tumor. Cancer. 1958; 11(3):455–459.Article Pubmed
[41]Speroni L, Bustuoabad Vde L, Gasparri J, Chiaramoni NS, Taira MC, et al. Alternative site of implantation affects tumor malignancy and metastatic potential in mice: Its comparison to the flank model. Cancer Biol Ther. 2009; 8(4):375–379.Article Pubmed
[42]Gorelik E, Segal S, Feldman M. Growth of a local tumor exerts a specific inhibitory effect on progression of lung metastases. Int J Cancer. 1978; 21(5):617–625.Article Pubmed
[43]Gorelik E, Segal S, Feldman M. Control of lung metastasis progression in mice: Role of growth kinetics of 3LL Lewis lung carcinoma and host immune reactivity. J Natl Cancer Inst. 1980; 65(6):1257–1264.Article Pubmed
[44]Greene HS, Harvey EK. The inhibitory influence of a transplanted hamster lymphoma on metastasis. Cancer Res. 1960; 20:1094–1100.Article Pubmed
[45]Gueron G, Anselmino N, Chiarella P, Ortiz EG, Lage Vickers S, et al. Game-changing restraint of Ros-damaged phenylalanine, upon tumor metastasis article. Cell Death Dis. 2018; 9(2):140.Article Pubmed
[46]Guba M, Cernaianu G, Koehl G, Geissler EK, Jauch KW, et al. A primary tumor promotes dormancy of solitary tumor cells before inhibiting angiogenesis. Cancer Res. 2001; 61(14):5575–5579.Article Pubmed
[47]Hamilton AM, Parkins KM, Murrell DH, Ronald JA, Foster PJ. Investigating the impact of a primary tumor on metastases and dormancy using MRI: New insights into the mechanisms of concomitant tumor resistance. Tomography. 2016; 2(2):79–84.Article Pubmed
[48]Ruggiero RA, Bruzzo J, Chiarella P, Di Gianni P, Isturiz MA, et al. Tyrosine isomers mediate the classical phenomenon of concomitant tumor resistance. Cancer Res. 2011; 71(22):7113–7124.Article Pubmed
[49]Strazza A, Montagna DR, Aixala M, Meiss RP, Chiarella P, et al. Revisiting the phenomenon of concomitant tumor resistance and its impact on established metastases of murine and human origin. Glob Adv Res J Med Med Sci. 2017; 6(12):2315–5159.Article
[50]Prehn RT. Two competing influences that may explain concomitant tumor resistance. Cancer Res. 1993; 53(14):3266–3269.Article Pubmed
[51]Ehrlich P. Experimentelle Carcinomstudien an mausen. Arbeiten aus dem Koiglichen Institut fur Experimentelle Therapie zu Frankfurt/ AM. 1906. 77-103 p.
[52]Bashford E, Murray J HM. General results of propagation of malignant new growths. In: E Bashford, editor. Third scientific report on the investigation of the Imperial Cancer Research Fund. London: Tay. 1908; 262–268.
[53]Woglom W. Immunity to transplantable tumors. Cancer Rev. 1929; 4:129–214.Article
[54]Southam C. Host defense mechanisms and human cancer. Ann Inst Pasteur (Paris). 1964; 107:585–597.Pubmed
[55]Gershon RK, Carter RL, Kondo K. On concomitant immunity in tumour-bearing hamsters. Nature. 1967; 213(5077):674–676.Article Pubmed
[56]Brunschwig A, Southam CM, Levin AG. Host resistance to cancer. Clinical experiments by homotransplants, autotransplants and admixture of autologous leucocytes. Ann Surg. 1965; 162(3):416–425.Article Pubmed
[57]Lausch R, Rapp F. Concomitant immunity in hamsters bearing DMBA-induced tumor transplants. Int J Cancer. 1969; 4(2):226–231.Article Pubmed
[58]Keller R. Repression of lymphatic metastasis by a second implant of the same tumor. Invasion Metastasis. 1985; 5(5):295–308. Pubmed
[59]Franco M, Bustuoabad OD, di Gianni PD, Goldman A, Pasqualini CD, et al. A serum-mediated mechanism for concomitant resistance shared by immunogenic and non-immunogenic murine tumours. Br J Cancer. 1996; 74(2):178–186.Article Pubmed
[60]Ando K, Hunter N, Lester J. Peters. Immunologically nonspecific enhancement of artificial lung metastases in tumor-bearing mice. Cancer Immunol Immunother. 1979; 6(3):151–156.Article
[61]Janik P, Bertram J SB. Modulation of lung tumor colony formation by a subcutaneously growing tumor. Natl Cancer Inst. 1981; 66(6):1155–1158.Article Pubmed
[62]McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008; 133(6):994–1005.Article Pubmed
[63]Elkabets M, Gifford AM, Scheel C, Nilsson B, Reinhardt F, et al. Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J Clin Invest. 2011; 121(2):784–799.Article Pubmed
[64]Parkins KM, Dubois VP, Hamilton AM, Makela AV, Ronald JA, et al. Multimodality cellular and molecular imaging of concomitant tumour enhancement in a syngeneic mouse model of breast cancer metastasis. Sci Rep. 2018; 8(1):8930.Article Pubmed
[65]Lokich J. Spontaneous regression of metastatic renal cancer. Case report and literature review. Am J Clin Oncol. 1997; 20(4):416–418.Article Pubmed
[66]Wyczolkowski M, Klima W, Bieda W, Walask K. Spontaneous regression of hepatic metastases after nephrectomy and mastectomy of renal cell carcinoma. Urol Int. 2001; 66(2):119–120.Article Pubmed
[67]Lekanidi K, Vlachou PA, Morgan B, Vasanthan S. Spontaneous regression of metastatic renal cell carcinoma: Case report. J Med Case Rep. 2007; 1:89.Article Pubmed
[68]Ray A, Ghosh SK. Long natural history and spontaneous regression: A rare outcome of metastatic renal cell carcinoma. Clin Cancer Investig J. 2014; 3(5):447–449.Article
[69]Bruzzo J, Chiarella P, Meiss RP, Ruggiero RA. Biphasic effect of a primary tumor on the growth of secondary tumor implants. J Cancer Res Clin Oncol. 2010; 136(10): 1605–1615.Article Pubmed
[70]North R. The murine antitumor immune response and its therapeutic manipulation. Adv Immunol. 1984; 35:89–155.Article Pubmed
[71]Dujardin-Beaumetz. Lecciones de Clínica Terapéutica. Madrid, España. 1880.
[72]O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, et al. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a lewis lung carcinoma. Cell. 1994; 79(2):315–328.Article Pubmed
[73]Ruggiero RA, Bustuoabad OD, Cramer P, Bonfil RD, Pasqualini CD. Correlation between seric antitumor activity and concomitant resistance in mice bearing nonimmunogenic tumors. Cancer Res. 1990; 50(22):7159–7165.Article Pubmed
[74]DeWys WD. Studies correlating the growth rate of a tumor and its metastases and providing evidence for tumor-related systemic growth-retarding factors. Cancer Res. 1972; 32(2):374–379.Article Pubmed
[75]Nakahara W, Fukuoka F. The newer concept of cancer toxin. Adv Cancer Res. 1958; 5:157–177.Article Pubmed
[76]Meiss RP, Bonfil RD, Ruggiero RA, Pasqualini CD. Histologic aspects of concomitant resistance induced by nonimmunogenic murine tumors. J Natl Cancer Inst. 1986; 76(6):1163–1175.Article Pubmed
[77]Franco M, Bustuoabad OD, di Gianni PD, Meiss RP, Vanzulli S, et al. Two different types of concomitant resistance induced by murine tumors: Morphological aspects and intrinsic mechanisms. Oncol Rep. 2000; 7(5):1053–1063.Article Pubmed
[78]Bertin C, Weston LA, Huang T, Jander G, Owens T, et al. Grass roots chemistry: Meta-Tyrosine, an herbicidal nonprotein amino acid. Proc Natl Acad Sci U S A. 2007; 104(43):16964–16969.Article Pubmed
[79]Gurer-Orhan H, Ercal N, Mare S, Pennathur S, Orhan H, et al. Misincorporation of free m-tyrosine into cellular proteins: A potential cytotoxic mechanism for oxidized amino acids. Biochem J. 2006; 395(2):277–284.Article Pubmed
[80]Machuca D, Chiarella P, Montagna D, Dran G, Meiss RP, et al. Meta-tyrosine. A powerful anti-metastatic factor with undetectable toxic-side effects. Medicina (B Aires). 2015; 75(1):1–5.Article Pubmed
[81]Ruggiero RA, Machuca D, Chiarella P, Gueron G, Vázquez E, et al. Tyrosine isomers mediate the classical phenomenon of concomitant tumor resistance and exhibit a powerful anti-metastatic effect with undetectable toxic-side effects. J Cancer Sci Ther. 2015; 7:8.Article
[82]Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006; 55(3):237–245.Article Pubmed
[83]Chiarella P, Reffo V, Bruzzo J, Bustuoabad OD, Ruggiero RA. Therapeutic anti-tumor vaccines: From tumor inhibition to enhancement. Clin Med Oncol. 2008; 2:237–245.Article Pubmed
[84]Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008; 111(12):5457–5466.Article Pubmed
[85]Zhang S, Ma X, Zhu C, Liu L, Wang G, et al. The role of myeloid-derived suppressor cells in patients with solid tumors: A meta-analysis. PLoS One. 2016; 11(10):e0164514.Article Pubmed
[86]Ruggiero RA, Bruzzo J, Chiarella P, Bustuoabad OD, Meiss RP, et al. Concomitant tumor resistance: The role of tyrosine isomers in the mechanisms of metastases control. 2012; 72(5):1043–1050.Article Pubmed
[87]Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012; 48(5):667–680.Article Pubmed
[88]Ling J, Yadavalli SS, Ibba M. Phenylalanyl-tRNA synthetase editing defects result in efficient mistranslation of phenylalanine codons as tyrosine. RNA. 2007; 13(11):1881–1886.Article Pubmed
[89]Man N, Chen Y, Zheng F, Zhou W, Wen LP. Induction of genuine autophagy by cationic lipids in mammalian cells. Autophagy. 2010; 6(4):449–454.Article Pubmed
[90]Ruggiero RA, Di Gianni PD, Franco M BO. Resistencia concomitante antitumoral. Med. 1996; 56(1):57–64.
[91]Coffey JC, Wang JH, Smith MJF, Bouchier-Hayes D, Cotter TG, et al. Excisional surgery for cancer cure: Therapy at a cost. Lancet Oncol. 2003; 4(12):760–768.Article Pubmed
[92]Jirtle RL, Michalopoulos G. Effects of partial hepatectomy on transplanted hepatocytes. Cancer Res. 1982; 42(8):3000–3004.Article Pubmed
[93]Joseph B, Berishvili E, Benten D, Kumaran V, Liponava E, et al. Isolated small intestinal segments support auxiliary livers with maintenance of hepatic functions. Nat Med. 2004; 10(7):749–753.Article Pubmed
[94]Glick AB, Yuspa SH. Tissue homeostasis and the control of the neoplastic phenotype in epithelial cancers. Semin Cancer Biol. 2005; 15(2):75–83.Article Pubmed
[95]Bustuoabad OD, Ruggiero AR. Hypothesis of the basic biological sense of cancer revisited. A putative explanation of Peto’s paradox. Cancer Hypothesis. 2017; 4(7):1–16.Article
[96]Prehn RT. The inhibition of tumor growth by tumor mass. Cancer Res. 1991; 51(1):2–4.Article Pubmed
 Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved