


- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Modern Human Pathology
Volume 3, Issue 3, April 2018, Pages 7–11
Case reportOpen Access
Primary osseous amyloidoma - Report of two cases with review of the literature
-
Clarissa Smith1,
Da Zhang1,
Larry Hoover2,
Howard Rosenthal3 and
Ossama Tawfik1,*

*Corresponding author: Ossama Tawfik, MD, PhD., Professor, Department of Pathology and Laboratory Medicine, The University of Kansas Medical Center, Kansas City, Kansas 66160, USA. Tel.: (913) 588-1185; E-mail: otawfik@kumc.edu
Received 05 February 2018 Revised 17 March 2018 Accepted 27 March 2018 Published 04 April 2018
DOI: http://dx.doi.org/10.14312/2397-6845.2018-3
Copyright: © 2018 Smith C, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
Amyloidoma, also known as tumoral amyloidosis, is a rare localized subset of amyloidosis without systemic involvement. Amyloid becomes aggregated in the extracellular matrix ofvarious tissues such as the respiratory, urinary, central nervous systems, heart, salivary glands, breast and orbital tissue. Very few cases of osseous amyloidosis have been reported in the literature, especially in the absence of systemic amyloidosis or dialysis. The two cases that follow describe such lesions which were identified in the bone.
Keywords: localized amyloidosis; osseous; histopathology; plasmacytoma
IntroductionTop
Most reported cases of primary osseous amyloidoma are localized in the axial skeleton, including eight published occurrences in the skull [1-8]. Appendicular involvement is far less common with only two cases reported in the humerus [9, 10] and two in the femur [11, 12]. We report two cases of isolated osseous amyloidomas: One in the anterior skull base and the other in the appendiceal skeleton. Both occurred in association with either a remote or recent history of a plasmacytoma. The clinicopathological and radiological findings of these two cases were reported with review of the literature discussing the importance of careful histopathologic evaluation for accurate diagnosis and management of these patients.
Case reportsTop
The first patient is a 62-year-old female who was referred to our oncologic orthopedic surgeon in reference to a lesion found in her right acetabulum. She reported to her primary care physician with moderate to severe dull aching pain of her right hip, increasing over the course of two months before admission. She was no longer fully weight-bearing and had been forced to modify her active lifestyle due to pain. The patient could not recall any inciting trauma.
The patient's past medical history was significant for a solitary IgA lambda-restricted plasmacytoma involving the right periacetabular region including roof, ischium, and ischial tuberosity that was discovered serendipitously during workup for unrelated complaint seventeen years prior to the current admission. After appropriate staging including bone marrow examination and definitive radiation therapy to the right hip, a complete remission was attained and the patient has been in remission since. Other relevant past medical history includes myomectomy in 1984, cesarean section in 1985, hysterectomy in 1992 and glaucoma in 2016.
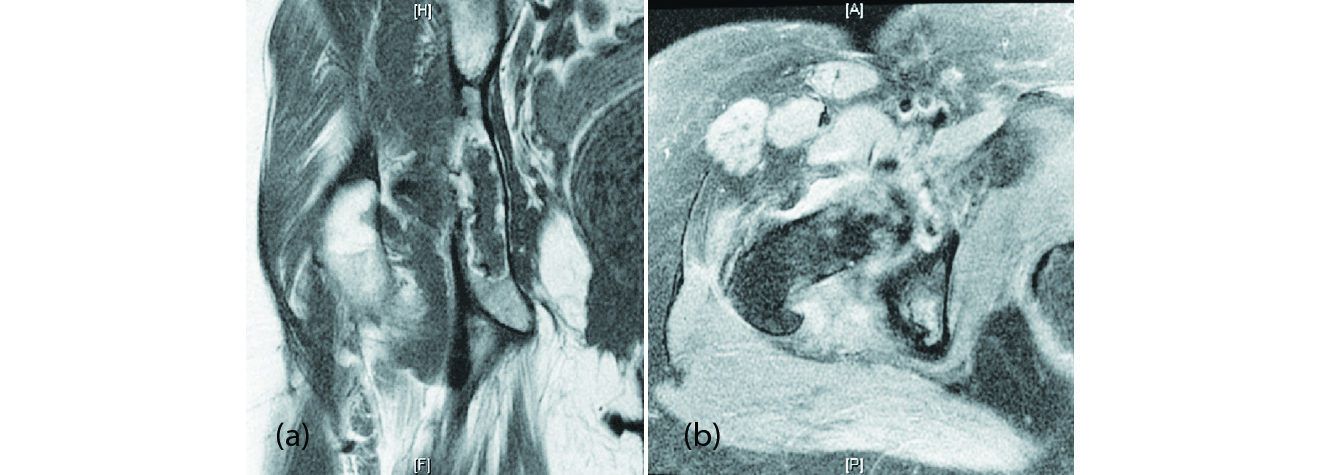
Examination of the right hip displayed no tenderness to palpation, no palpable mass, no overlying skin changes and a full range of motion without pain. The rest of the physical examination was unremarkable. Radiologic studies including x-ray and MRI revealed a large cystic lytic defect involving the right hip area that was concerning for a recurrence of her plasmacytoma or other neoplastic processes. Magnetic resonance imaging (MRI) findings included diffuse abnormal signal intensity identified within the right acetabulum (Figure 1). Additional findings included mild degenerative changes with loss of the articular cartilage involving the superior aspect of the right hip, subarticular cystic change and edema identified in the ear aspect of the acetabulum, synovitis, as well as contrast enhancement and increased STIR signal intensity within the pectineus and obturator externus muscles suggestive of myositis. Alesional biopsy was performed showing polyclonal plasma cells with no evidence of plasma cell dyscrasia. Blood counts and electrophoresis of urine and serum indicated antibody production was within normal limits. Given the persistence of her pain over a lengthy period, an open biopsy followed by intralesional curettage and bone grafting was recommended. In addition to the diagnostic utility of this approach, it would also provide pain reduction and a more mechanically sound foundation for future arthroplasty, which this patient has an elevated risk of needing.
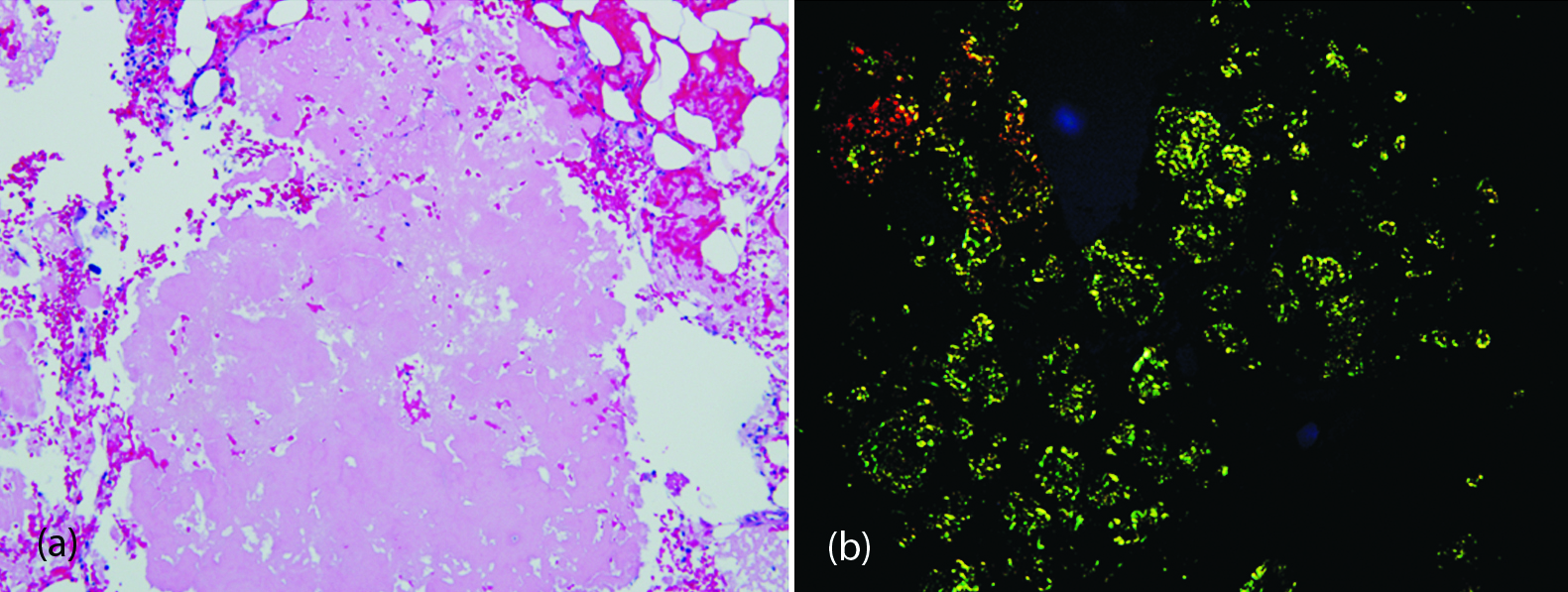
Intraoperative biopsy specimens received by the surgical pathology lab were ‘aggregates of red-brown soft tissue fragments’. Microscopic observations of frozen sections were called to the operating room as notable for necrosis without evidence of neoplasia. Further observation of the submitted tissues revealed abundant proteinaceous deposits remarkable for apple green birefringence with Congo Redamyloid stain, indicative of amyloid deposits (Figure 2). The bone marrow showed reactive change without apparent increase of plasma cells. Immunostains to rule out metastatic disease including carcinomas and melanoma were negative. Flow cytometry confirmed the lack of increased plasma cells and indicated polyclonality of those present. Fluorescent in situ hybridization studies confirmed the polyclonality of plasma cells.
Two weeks following her procedure the patient tripped and stepped down hard onto her right hip experiencing right hip pain. An x-ray was performed showing no signs of fracture and the redemonstration of the lytic lesion within the right acetabulum representing either partial incorporation of the bone graft or persistence of residual disease. A subsequent follow up bone marrow biopsy two weeks later was performed showing a normocellular (40%) marrow with trilineage hematopoiesis and 6% polyclonal plasma cells without evidence of multiple myeloma/plasma cell dyscrasia. Currently the patient is being followed up by her hematologist with no further treatment.
The second case is that of a 70-year-old man who presented to our otolaryngology clinic with recurrent sinus infections involving maxillary and frontal sinuses, which were increasing in frequency and severity over the last five years, despite intermittent self-administered irrigations and topical steroids. A balloon sinuplasty was performed in 2015 at an outside institution to manage these problems, but the patient’s painful infections returned shortly after. Social history also reveals a 12.5 pack-year smoking history.
Initial nasal endoscopic examination revealed an edematous sinus mucosa without apparent polyps, polypoid degeneration, retained secretions, or purulence. Accordingly, the patient was started on regular topical nasal steroid drops and saline nasal irrigations. LandmarX CT sinus scan demonstrated a mass-like polypoid mucosal thickening in the left posterior nasal cavity and sphenoid region of the skull base. The lobulated morphology and shiny smooth surface of the mass increased concern for a primary neoplasm such as a nasopharyngeal carcinoma (Figure3). Several soft, red-brown tissue fragments were removed and submitted to the surgical pathology lab in formalin, which revealed respiratory mucosa along with acellular extracellular material, consistent with amyloid. A Congo red stain was performed to confirm the diagnosis. Androgen receptor and CD34 staining were also performed to rule out nasopharyngeal angiofibroma.
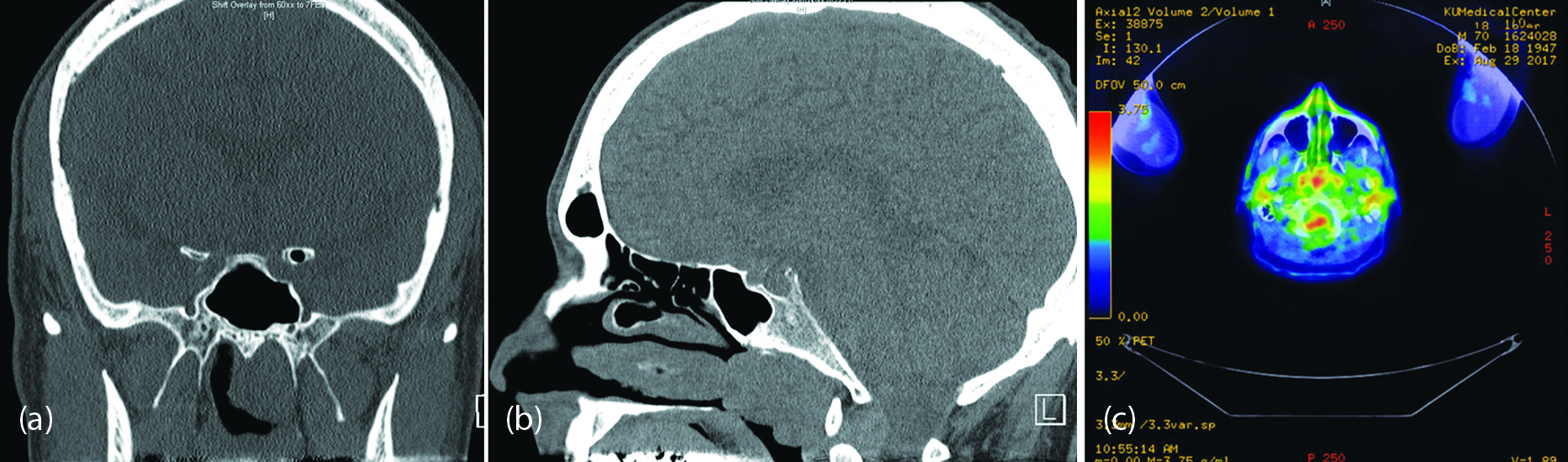
To investigate the origin of the amyloid, the patient was referred to oncology. Bone marrow biopsy, skeletal survey, and extensive testing of urine and blood were ordered due to the association of amyloid with lymphocytic malignancies such as lymphoma and plasma cell dyscrasia. Because the amyloidoma described in the first case had recently been discussed in the pathology department, the idea of a solitary amyloid aggregation was also suggested as a possible diagnosis. The patient’s serum exhibited normal levels of immunoglobulin’s and no monoclonality by electrophoresis. While the total protein of the urine was slightly high at 187mg/mmol, electrophoresis with immunofixation was negative for paraprotein as well as for elevated globulins or monoclonality. Circulating lymphocytes appeared normal in number and morphology. Bone marrow biopsy and aspirate from the iliac crest demonstrated quantitatively normal kappa and lambda light chains and a low-normal ratio of 0.3. Flow cytometry indicated a minor (4%) population of monoclonal CD5 lambda-restricted B lymphocytes, which prevented elimination of B-cell lymphoma or monoclonal B-cell lymphocytosis from the differential diagnosis. Therefore, a PET scan was ordered to look for other active lesions. By PET, the nasopharyngeal mass demonstrated low-level uptake, which could represent an inflammatory process or low-grade neoplasm, such as lymphoma. Radiology recommended further characterization by tissue sampling. Skeletal survey also did not demonstrate any suspicious lytic or blastic lesions.
A Functional Endoscopic Sinus Surgery (FESS) was performed under LandmarX guidance to restore the airway, open chronically infected maxillary and ethmoid sinuses, and excise the remaining nasopharyngeal mass. Bilateral maxillary antrostomy, bilateral anterior ethmoidectomy, and inferior middle turbinate trim drained the infected sinuses and enabled better access to remove the lesion with biting instruments and suction debriders. Pathology of removed maxillary and ethmoid sinus tissue showed chronic sinusitis without eosinophilia. Microscopic examination of the nasopharyngeal mass was notable for amyloid with scattered monoclonal lambda-restricted plasma cells making up less than 5% of the specimen.
DiscussionTop
Amyloidoma of bone is an uncommon finding, especially in the absence of primary systemic amyloidosis or dialysis. This tumoral form of amyloidosis characterized by the localized deposition of amyloid is a diagnostic challenge. Review of the literature reveals the overwhelming majority of skeletal amyloidoma occur in axial skeleton loci such as the spine, skull, and ribs [11]. Of the two amyloidoma cases we reported, one occurred in the axial and the other in the appendiceal skeleton. This lesion’s unusual presence in the appendicular skeleton is of particular interest, as it may bolster the correlation with the preceding osseous plasmacytoma. Additional data suggest that osseous plasmacytoma located in the axial skeleton are associated with increased risk for recurrence [14]. Interestingly, the patient with history of the appendicular lesion is still free from recurrence or progression, seventeen years after her primary diagnosis.
Deposition of amyloid is often a diffuse or multi-centric process as a function of its origination from translational or post-translational derangements causing rampantly aberrant quaternary structure. It is the result of a phenomenon in which low molecular weight proteins undergo erroneous misfolding events leading to insoluble fibrillated aggregates of anti-parallel beta pleated sheets [15]. These frequently become deposited in various tissues and contribute to the wide range of clinical presentations associated with this group of illnesses. While this general pathophysiological schema is similar among cases in the literature, several variables make this group of diseases diagnostically challenging. For one, there are a significant number of proteins which have been implicated in the formation of amyloid, and the list continues to grow with uncommon variants. In their normal conformations, most of these are small soluble subunits crucial to structures with essential physiologic functions [15]. Another variable contributing to the diagnostic challenge is the diversity of presentation. If left untreated, amyloid can deposit in nearly any organ, leading to its progressive dysfunction and eventual failure.
Due to the complex characteristics of amyloid, there are multiple ways to categorize the group of diseases known as amyloidoses. Although many instances represent systemic pathophysiology, both cases presented above are apparently restricted to a singular location. Thus, the term amyloidoma or tumoral amyloidosis provides a useful description of this entity. Another common classification scheme used throughout related literature is primary versus secondary. These correspond to the two most frequently encountered types of amyloid which are AL and AA. The fibrillating protein in AL amyloid is the lambda light chain, which explains its frequent association with plasma cell dyscrasias [14]. Conversely, the aggregating protein in AA, or secondary, amyloid is a protein elevated during states of prolonged inflammation known as serum amyloid protein A [15].
In general, amyloid cannot be definitively categorized as primary or secondary amyloid without further testing by mass spectrometry or additional immunostains. The amyloidomas in both patients are presumed to be comprised of the AL, or primary variant, due to the patients’ history of plasmacytoma in the same location. Interestingly, they seem to be in the minority of patients who do not progress to multiple myeloma within the first three years [15].
Amyloidosis is relatively easy to identify when clinical suspicion is high. With Congo red stain, the proteinaceous aggregates exhibit a characteristic apple green birefringence under polarized light [16]. Often, the protein contributing to the pathology is identifiable in circulation through serology, or the patient’s past medical or family history will give an insight. In the absence of these clues, however, atypical presentations as demonstrated in the cases above are especially challenging to manage. Identifying, or at the very least, ruling out certain underlying causes is crucial to optimal outcomes for patients with amyloidoma.
Following a thorough history, imaging and biopsy of the lesion are important sources of early information. In both cases, the lesion was described as cystic and lytic by radiology, raising suspicion for neoplasms such as chondrosarcoma. Biopsy, however, established the diagnosis of amyloidoma. Once amyloid is identified, the diagnostic process shifts to ruling out sinister underlying causes. Blood and urine electrophoresis with immunofixation can provide quantitative and qualitative data about proteins with inflammatory and immunological functions. Renal function should also be serologically investigated, as the kidneys are a site frequently impaired by systemic amyloid. It is important to note, however that finding a monoclonal protein in the serum is not definitive for AL amyloid. One study suggests that up to 10% of cases exhibiting a monoclonal serum or urine protein are due to some other protein misfolding [17]. Analysis of bone marrow by core biopsy and aspirate can also elucidate correlative relationships in many instances. Specimen should be carefully analyzed with immunostains and flow cytometry for conditions which could result in an excess of light chains, such as multiple myeloma and lymphoma.
ConclusionTop
In conclusion, amyloidoma is an important clinical entity to recognize because it is often the herald of a greater underlying issue. The process of protein misfolding can indicate alterations at the level of genetic code, deleterious post-translational events, or a deficiency of chaperone-like proteins which help correct occasional spontaneous derangements [18]. Often, however, the misfolding and aggregation is secondary to significantly increased quantities of certain low molecular weight proteins. This excess can indicate processes in which regulation is deranged like neoplasia, or some type of failure to clear a normal quantity of protein product such as organ dysfunction or dialysis. Understanding the process of amyloid formation and deposition provides valuable insight for thorough investigation of its insidious underlying causes.
Conflict of interest
Authors declare no conflicts of interest related to this case report.
ReferencesTop
[1]Giordano A, Horne DG, Gudbrandsson F, Meyerhoff W. Temporal bone amyloidoma. Otolaryngol Head Neck Surg. 1983; 91(1):104–108.Article Pubmed
[2]Ferreiro JA, Bhuta S, Nieberg RK, Verity MA. Amyloidoma of the skull base. Arch Pathol Lab Med. 1990; 114(9):974–976.Pubmed
[3]Unal F, Hepgül K, Bayindir C, Bilge T, Imer M, et al. Skull base amyloidoma. Case report.J Neurosurg.1992; 76(2):303–306.Article Pubmed
[4]Hidalgo F, Aguilera C, Monfort JL, Reñé M, Muntané A, et al. Amyloidoma of the skull: Plain radiographs, CT and MRI. Neuroradiology.1996; 38(1):44–46.Article Pubmed
[5]Simoens WA, van den Hauwe L, Van Hedent E, Warson F, Demaeseneer R, et al. Amyloidoma of the skull base. Am J Neuroradiol.2000; 21(8):1559–1562.Article Pubmed
[6]Oruckaptan H, Karli Oğuz K, Işikay I, Ruacan S. Amyloidoma of the temporal bone and upper cervical spine; presentation of a rare clinical entity with a brief literature review. Turk Neurosurg.2009; 19(2):159–162.Article Pubmed
[7]Parmar H, Rath T, Castillo M, Gandhi D. Imaging of focal amyloid depositions in the head, neck, and spine: Amyloidoma. Am J Neuroradiol.2010; 31(7):1165–1170.Article Pubmed
[8]Agrawal M, Batra VV, Majumdar K, Upreti L, Singh D. Extensive osteolytic skull base amyloidoma simulating malignancy: A rare pseudotumor producing a diagnostic dilemma. J Cancer Res Ther.2015; 11(3):646.Pubmed
[9]Iguchi T, Kizaki M, Kurauchi A, Yano T, Ikeda Y. Soft tissue amyloidoma. Rinshō ketsueki.2005; 46(7):507–512.Article Pubmed
[10]Pambuccian SE, Horyd DI, Cawte T, Huvos AG. Amyloidoma of bone, a plasma cell/plasmacytoid neoplasm: Report of three cases and review of the literature. Am J SurgPathol.1997; 21(2):179–186.Article Pubmed
[11]Verhoeven F, Prati C, Wendling, D. Amyloidoma, an unusual cause of fracture. Case Rep Rheumatol..2014; 2014:424056.Article Pubmed
[12]Lai KN, Chan KW, Siu DL, Wong CC, Yeung D. Pathologic hip fractures secondary to amyloidoma. Case report and review of the literature. Am J Med. 1984; 77(5):937–943.Article Pubmed
[13]Bataille R, Sany J. Solitary myeloma: Clinical and prognostic features of a review of 114 cases. Cancer.1981; 48(3):845–851.Article Pubmed
[14]Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, et al. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. J Protein Fold Disorders.2016; 23(4):209–213.Article Pubmed
[15]Dimopoulos MA, Goldstein J, Fuller L, Delasalle K, Alexanian R. Curability of solitary bone plasmacytoma. J ClinOncol.1992; 10(4):587–590.Article Pubmed
[16]Howie AJ, Brewer DB. Optical properties of amyloid stained by Congo red: History and mechanisms. Micron.2009; 40(3):285–301.Article Pubmed
[17]Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002; 346(23):1786–1791.Article Pubmed
[18]Stefan M, Rigacci S. Protein folding and aggregation into amyloid: The interference by natural phenolic compounds. Int J Mol Sci. 2013; 14(6):12411–12457.Article Pubmed
 Copyright
© 2012-2025 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2025 NobleResearch Group. All Rights Reserved