



- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Clinical Radiation Oncology
Volume 1, Issue 3, September 2016, Pages 13–15
Case reportOpen Access
Malignant peripheral nerve sheath tumor: Second or secondary malignancy after Hodgkin’s lymphoma treatment in childhood
-
Agata Adamczewska1,*
 ,
Julita Janura–Szymańska1,
Agnieszka Wziątek1,
Jacek Wachowiak1and
Katarzyna Derwich1
,
Julita Janura–Szymańska1,
Agnieszka Wziątek1,
Jacek Wachowiak1and
Katarzyna Derwich1
*Corresponding author: Agata Adamczewska, MD, Department of Pediatric Oncology, Hematology and Transplantology, Poznan University of Medical Sciences, Poznań, 60-572, Poland. E-mail: aadamczewska@skp.ump.edu.pl
Received 15 May 2016 Revised 10 August 2016 Accepted 23 August 2016 Published 30 August 2016
DOI: http://dx.doi.org/10.14312/2397-8511.2016-3
Copyright: © 2016 Adamczewska A, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
Malignant peripheral nerve sheath tumour (MPNST) is a very rare soft tissue tumour with poor prognosis. The article describes the case of a patient cured from Hodgkin’s lymphoma (HL) by means of chemotherapy and radiotherapy who developed the mediastinal tumour histopathologically corresponding to MPNST 10 years after completion of the therapy.
Keywords: chemotherapy; children; Hodgkin’s lymphoma; radiotherapy; secondary malignancy; soft tissue sarcoma; triton tumour
IntroductionTop
Malignant peripheral nerve sheath tumour (MPNST) is a rare spindle cell tumour belonging to the class of soft tissue sarcomas usually occurs de novo [1]. The risk factors are neurofibromatosis type 1 (NF1) and prior radiation therapy (in ca. 11% of cases MPNST) [2]. The primary method of treatment is radical tumour resection followed by adjuvant radiation therapy [1]. Multidrug chemotherapy is not routinely recommended. [2] MPNST is associated with a very poor prognosis: the mean survival time is around 30-45 months [1].
Case reportTop
A 15-year-old boy was diagnosed with a tumour in the upper left mediastinum, histopathologically corresponding to MPNST, with no distant metastases. At the age of 5 years the patient was diagnosed with HL, classical type, stage IIA and treated with chemotherapy (three cycles of B-DOPA: bleomycin 4 mg/m2 intravenous (i.v.) day 2, 5, dacarbazine 150 mg/m2 i.v. day 1-5, vincristine 1,5 mg/m2 i.v. day 1, adriamycin 60 mg/m2 i.v. day 1, 5, prednisone 40 mg/m2 p.o. day 1-5 and two cycles of MVPP: nitrogranulogen 6 mg/m2 i.v. day 1, 8, vinblastine 6 mg/m2 i.v. day 1, 8, procarbazin 100 mg/m2 p.o. day 1 - 10, prednisone 40 mg/m2 p.o. day 1-10) and irradiation (dose of 25 Gy/T to the cervical/supraclavicular lymph node region) according to the protocol of the Polish Paediatric Leukemia/ Lymphoma Study Group – HD 97 [3].
Ten years after completing HL treatment the patient was diagnosed with left-sided Horner’s syndrome. An X-ray examination of the chest revealed opacity within the apex of the left lung, with a polycyclic lower border-visible oval-shaped, partially well-circumscribed shadows (one measuring 5.6 × 3.8 cm), probably consistent with a solid lesion. The features of the findings suggest a hyperplastic lesion with accompanying inflammatory/ atelectatic lesions in segment LS 1/2. An additional CT scan of the chest was performed, yielding the following findings: a well-circumscribed heterogeneous (with density varying from 15 to 70 jH) nodular mass measuring 8.2 × 7.5 cm (ds × ap-measurements in axial scans) × 5.6 cm (cc) visible in the upper posterior mediastinum on the left. The lesion reaches the superior thoracic aperture, and posteriorly adheres to the vertebral bodies as well as posterior and lateral sections of the four upper ribs, and also adjoins the trachea and the oesophagus. The mass surrounds and deforms the initial portions of the left common carotid artery, the left internal jugular vein and left subclavian vessels, and adheres to the upper circumference of the aortic arch (Figure 1).
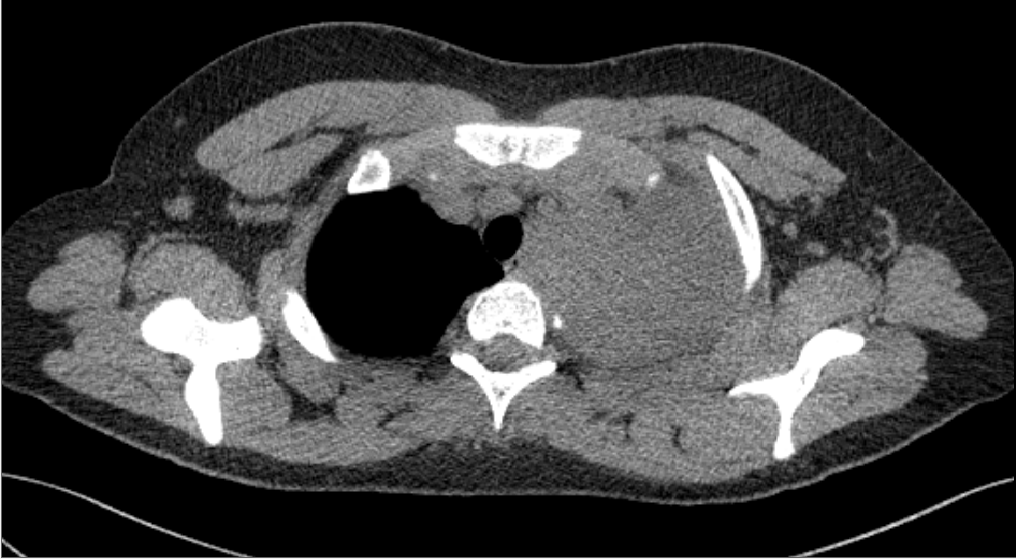
Tumour biopsy was performed twice. A needle biopsy failed to provide a diagnostic result, and a subsequent open biopsy yielded the diagnosis of neurofibroma/ ganglioneurofibroma. Due to the clinical progression of the disease, growing muscular weakness in the left hand and tumour mass enlargement to 13.3 × 10.3 cm seen on an imaging test, a third operative procedure was performed (non-radical tumour resection), based on which MPNST was diagnosed. Tumour cells demonstrated the presence of S-100 protein, neurofilament and neuron-specific enolase, in a small number of cells – B-cell lymphoma, p53, with a negative reaction for epithelial membrane antigen, desmin and smooth muscle actin. Around 70-80% of cells were positive for Ki-67. The boy was identified with unfavourable factors: large amount of tumour necrosis and high mitotic index. NF1 was excluded.
In view of the location and extent of the lesion it was impossible to perform a primary radical surgical procedure, so the patient began chemotherapy according to the CWS 2006 protocol for the high-risk “non-RMS-like” group (ifosfamide, vincristine, actinomycin, adriamycin) [4]. The chemotherapy regimen failed to induce any tumour mass reduction. The patient was then accepted for second-line chemotherapy according to CWS 2006 (carboplatin, etoposide, ifosfamide). After seven weeks of second-line chemotherapy a MRI scan showed the tumour to be unchanged. At the same time, the severity of the clinical symptoms of the disease increased, and the patient started to experience pain in the left hand. Non-standard chemotherapy with vincristine and cyclophosphamide was introduced. Overall, the patient received six cycles of chemotherapy and radiation therapy (total dose: 50.4 Gy administered to the mediastinal region). Imaging scans of the chest failed to show any signs of tumour mass reduction [4, 5].
Massive disease progression was seen 19 months after the primary diagnosis. Imaging scans showed a lesion, around 5 cm in size, located within the spinal canal, in the Th1-Th3 section, compressing the spinal cord and causing its posterior displacement. Palliative chemotherapy (second-line chemotherapy according to CWS-2002-P: 4 cycles of the topotecan 1 mg/m2 i.v. day 1-4, carboplatin 150 mg/m2 i.v. day 1-4) and anti-oedematous treatment (mannitol, steroids) were administered. [5] Palliative decompression of the spinal cord in the cervical section was performed. Tissue samples confirmed the diagnosis of MPNST with rhabdomyomatous differentiation (triton tumour G3) (Figure 2). The patient died 25 months after MPNST diagnosis.
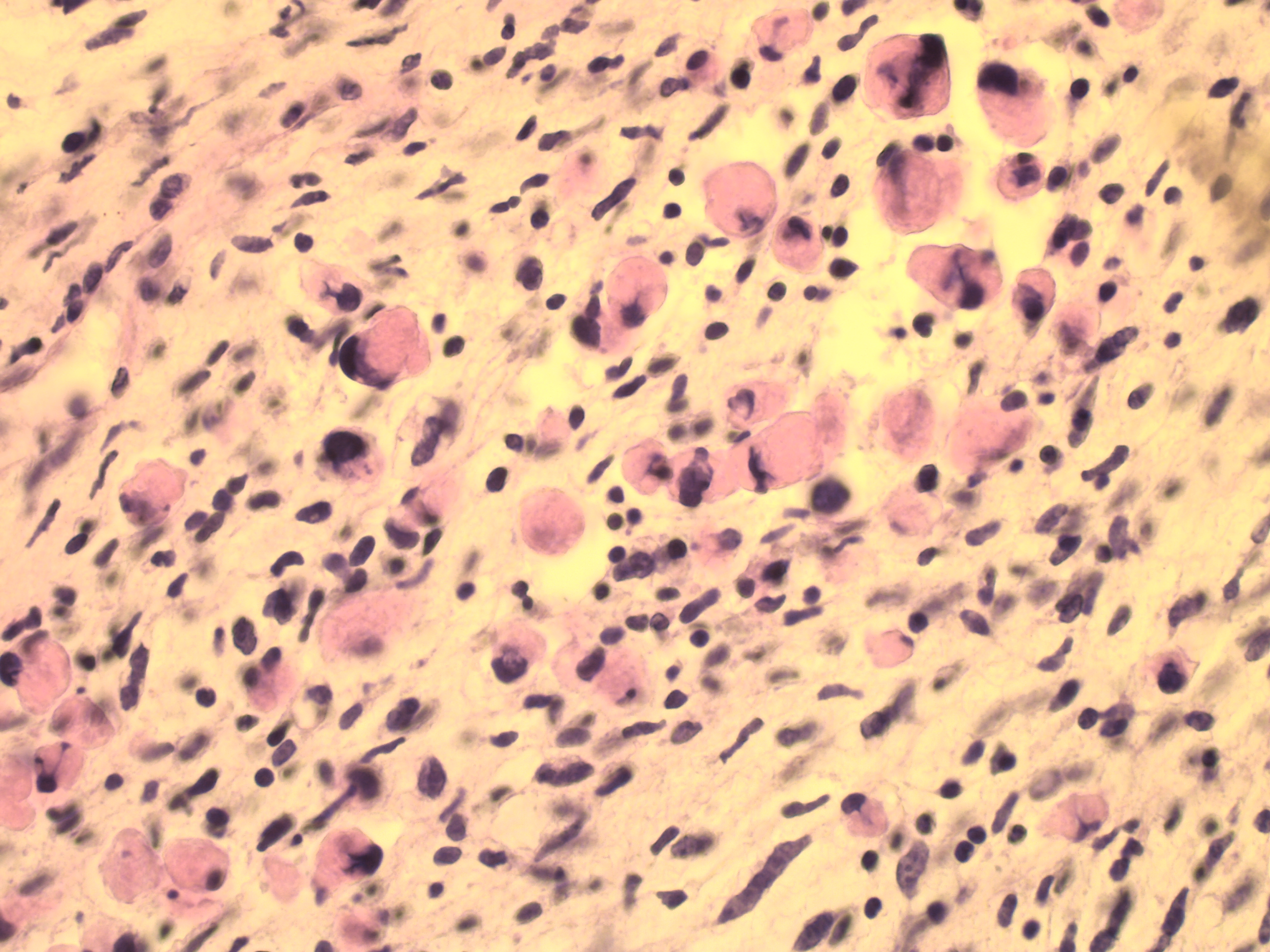
DiscussionTop
MPNST is a very rare soft tissue tumour with an incidence in children estimated to be under 0.001%. A major importance in MPNST epidemiology is attributed to NF1 and to prior radiotherapy if occurring as a secondary malignancy. Radiation therapy is a known etiological factor triggering secondary tumours. The most common of them are osteosarcoma and fibrosarcoma. There are case reports of MPNST developing as a secondary tumour after radiation therapy. According to Ducatman et al., out of 120 diagnosed cases of MPNST 11% were irradiation-associated, especially in NF1 patients [6]. Radiation therapy administered in combination with chemotherapy, particularly in the treatment of HL, may contribute to the development of MPNST as a secondary tumour. The literature describes the case of a patient who developed MPNST (mediastinal tumour) nine years after HL diagnosis. Despite undergoing surgery and chemotherapy, the patient died within two years of MPNST diagnosis [7]. Another patient was diagnosed with metastatic MPNST originating in the left brachial plexus. The disease occurred 10 years after radiation therapy for HL. The patient died 18 months after the diagnosis [8]. The mean period from radiation therapy to the development of a secondary tumour is estimated to be 15.6 years [9]. Also, attention should be given to another triggering factor, i.e. frequently performed imaging examinations (particularly CT scans) and their potential effect on carcinogenesis in children. Oncological patients, particularly diagnosed with solid tumours, require diagnostic imaging procedures both before establishing the final diagnosis and commencing therapy and during the evaluation of treatment efficacy and monitoring of disease remission. According to available estimates, out of 600,000 CT scans of the head and abdomen which are performed in the USA each year in patients under the age of 15, a total of 500 patients may develop a tumour related to exposure to harmful radiation [10].
Secondary tumours are also a major late complication of chemotherapy and affect 10% of all patients, though they can occur in 15% of patients with a history of HL within 20 years of diagnosis [11, 12]. If radiation therapy during the primary HL treatment had been reduced to an absolute minimum, perhaps it might have been possible to prevent MPNST in the patient discussed in the present study. According to the Polish Paediatric Leukemia/ Lymphoma Study Group – HD 97 protocol, irradiation was administered to all patients at disease stages IA HR, IB, IIA HR, IIB, III and IV [3]. At present, in conformity with the EuroNet PHL C1 protocol which is followed in Poland, the decision to commence radiation therapy is not taken until the completion of two initial chemotherapy cycles [13]. Irradiation is administered only to those patients whose response to therapy is unsatisfactory. This approach makes it possible to avoid radiation therapy even in patients with stage IV disease (provided that they have a good response to chemotherapy).
Moreover after HL treatment there is also a risk of developing a second malignancy with relative risk 4,6/ 10,000 person-years. The relative risk is higher with combined chemotherapy and radiotherapy than with radiation therapy alone (4,1 vs 6,0) [14].
The patient described above received chemotherapy (including nitrogranulogen) and radiation therapy for HL at a pre-school age. Ten years later, he was diagnosed with MPNST located in the upper left mediastinum, probably as a second or secondary malignancy. Due to the significant correlation existing between the occurrence of MPNST and NF1, despite the absence of clinical manifestations of neurofibromatosis, the patient was examined to determine possible rearrangements in the 17q11.2 region, however the result was negative.
MPNST is characterized by a very poor prognosis. The basic treatment method is radical surgical resection followed by radiation therapy. Since radical tumour resection was impossible throughout the entire treatment period, the patient discussed here received successive chemotherapy cycles and radiation therapy. Irradiation treatment was delayed by around two months due to concerns about further secondary carcinogenesis. The inefficacy of treatment and the observed gradual progression of the disease during chemotherapy corroborated the resistance of MPNST to chemotherapy and radiation therapy which has been reported in the literature [15]. In addition, the boy was identified with a number of factors which are known to have an adverse effect on prognosis – including prior irradiation treatment, large amount of tumour necrosis, high mitotic index and disease progression during therapy. It must also be noted that the repeated histopathological analysis diagnosed Triton tumour which is an extremely rare form of MPNST with a high malignancy index.
ConclusionTop
Summing up, a patient with a history of HL treatment (chemotherapy, radiation therapy) and multiple imaging examinations (X-ray and CT scans of the chest) developed with high probability a secondary tumour (MPNST) within the irradiated site, i.e. in the upper left mediastinum. Since radical resection was not a viable option because of poor accessibility of the tumour, extremely malignant nature of MPNST and presence of poor prognostic factors, multidrug chemotherapy (four lines of treatment in total) and radiation therapy were administered. The treatment failed to yield the desired effect. Taking into account the poor outcome of treatment in the patient discussed, restricting anticancer therapy to a required minimum with a reduction or elimination of radiation therapy, coupled with treatment adjustment to the stage of the disease, could make it possible in the future to reduce late side effects, including a secondary malignancy, associated with the treatment of primary tumours in childhood.
Funding
The article is financed from statutory research number 502-01-01104119-04448.
Conflicts of interests
Authors declare no conflicts of interest.
ReferencesTop
[1]Bates JE, Peterson CR, Dhakal S, Giampoli EJ, Constine LS. Malignant peripheral nerve sheath tumors (MPNST): a SEER analysis of incidence across the age spectrum and therapeutic interventions in the pediatric population. Pediatr Blood Cancer. 2014; 61(11):1955–1960.Article Pubmed
[2]Tuziak T, Dębiec-Rychter M, Zdzienicki M. Złośliwy nowotwór wywodzący się z osłonek nerwów obwodowych (malignant peripheral nerve sheath tumor, MPNST) In: Rutkowski T, Nowecki Z. Mięsaki tkanek miękkich). Medical Tribune. 2009.Article
[3]Balwierz W, Armata J, Moryl-Bujakowska A. Hodgkin’s lymphoma paediatric treatment regimen modified by Polish Paediatric Leukaemia/Lymphoma Study Group – HD 97 HD-97. 1996.
[4]Koscielniak E, Klingebiel T. Cooperative Weichteilsarkom Studiengruppe CWS der GPOH. Polish modification by Polish Paediatric Leukaemia/Lymphoma Study Group, CWS-2006. 2006.
[5]Cooperative Weichteilsarkom Studiengruppe CWS der GPOH. Polish modification by Polish Paediatric Leukaemia/Lymphoma Study Group, CWS-2002 P. 2002.
[6]Ducatman B, Scheithauer B, Piepgras D, Reiman H, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986; 57(10):2006–2021.Article Pubmed
[7]Simon Z, Ress Z, Toldi J, Trauninger A, Miltényi Z, et al. Rare association of Hodgkin lymphoma, Graves’ disease and myasthenia gravis complicated by post-radiation neurofibrosarcoma: coincidence or genetic susceptibility? Intl J Hematol. 2009; 89(4):523–528.Article Pubmed
[8]Fenzi F, Moretto G, Zamboni G, Passarin MG, Rizzuto N. Brain metastases from post-radiation malignant peripheral nerve sheath tumour. Ital J Neurol Sci. 1995; 16(6):495–498.Article Pubmed
[9]Ducatman B, Scheithauer B. Postirradiation neurofibrosarcoma. Cancer. 1983; 51(6):1028–1033.Article Pubmed
[10]Brenner D, Elliston C, Hall E, Berdon W. Estimated risks of radiation-induced fatal cancer from pediatric CT. AJR Am J Roentgenol. 2001; 176(2):289–296.Article Pubmed
[11]Kowalczyk JR, Samardakiewicz M. Rola pediatry pierwszego kontaktu w opiece nad dzieckiem przewlekle chorym, Odcinek 7: Choroba nowotworowa.Article
[12]Nyandoto P, Muhonen T, Joensuu H. Second cancer among long-term survivors from Hodgkin's disease. Int J Radiat Oncol Biol Phys. 1998; 42(2):373–378.Article Pubmed
[13]Korholz D, Wallace W, Landmann-Parker J. First inetrnational inter-group study for classical Hodgkin’s lymphoma in children and adolescents. EuroNet-Paediatric Hodgkin’s Lymphoma Group. 2012; 1–210.Article
[14]Ng A, Bernardo P, Weller E, Backstrand K, Silver B. Second malignancy after Hodgkin disease treated with radiation therapy with or without chemotherapy: long-term risks and risk factor. Blood. 2002; 100(6):1989–1996.Article Pubmed
[15]Kar M, Deo SV, Shukla NK, Malik A, DattaGupta S, et al. Malignant peripheral nerve sheath tumors (MPNST) – clinicopathological study and treatment outcome of twenty-four cases. Worl J Surg Oncol. 2006; 4:55.Article Pubmed
 Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved