



- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Diabetology and Endocrinology
Volume 2, Issue 1, July 2017, Pages 1–10
Original researchOpen Access
Long-term correction of diabetic hyperglycemia through glucose-responsive hepatic insulin production using lentivirus
-
Andrew M. Handorf1,
Hans W. Sollinger1,*
 and
Tausif Alam1
and
Tausif Alam1
*Corresponding author: Hans W. Sollinger, Department of Surgery, Division of Transplantation, University of Wisconsin-Madison, 600 Highland Ave., BX7375 CSC-H4, Madison, WI, 53792, USA. Tel.: (608) 263-9903; Fax: (608) 262-6280; E-mail: hans@surgery.wisc.edu
Received 04 April 2017 Revised 06 June 2017 Accepted 20 June 2017 Published 27 June 2017
DOI: http://dx.doi.org/10.14312/2398-0281.2017-1
Copyright: © 2017 Handorf AM, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
Type 1 diabetes mellitus (T1DM) is caused by the autoimmune destruction of the insulin-producing β cells of the pancreas. Insulin gene therapy is a promising strategy capable of overcoming the limitations of current treatments, but to become a viable option, it must provide long-term, glucose-responsive control of insulin production. We have previously achieved glucose-responsivity by incorporating glucose-inducible response elements (GIREs) upstream of a liver-specific insulin expression cassette (3xGIRE.ALB.Ins1-2xfur). In this study, 3xGIRE.ALB.Ins1-2xfur was delivered into streptozotocin-induced diabetic rats using lentivirus, resulting in remission of diabetic hyperglycemia for at least 482 days while restoring rate of weight gain in a dose-dependent fashion. Insulin immunostaining showed abundant insulin production in the liver, and qPCR showed 13-20 lentiviral integrations per cell in the liver of rats treated with high dose lentivirus. Negligible integration was found in the pancreas, kidney, spleen and muscle of LV-treated rats, confirming liver specificity. In vitro, LV.3xGIRE.ALB.Ins1-2xfur produced a 4.5-fold increase in insulin production in high glucose conditions, and in vivo, a 1.7-fold increase in insulin levels was found during an intraperitoneal glucose tolerance test. Unfortunately, limitations in large-scale lentivirus production and use of a tissue-specific promoter prevented treatment of more than one rat per batch of lentivirus. Thus, two of the LV-treated diabetic rats were undertreated, while another two rats were over treated, becoming hypoglycemic in the fed state. Nonetheless, we have established the framework for a long-term, glucose-responsive treatment for T1DM from which further improvements can be made.
Keywords: gene therapy; insulin; type 1 diabetes mellitus; lentivirus; transdifferentiation; long-term glucose control
IntroductionTop
Type 1 diabetes mellitus (T1DM) is a chronic metabolic disorder resulting from the autoimmune destruction of the insulin-producing β cells of the pancreas. The predominant treatment option requires multiple daily insulin injections to control hyperglycemia, but normal glucose regulation remains elusive. As a result, exogenous insulin therapy delays, but does not prevent, the onset of secondary complications associated with T1DM. Whole pancreas and islet transplantation therapies result in better glucose regulation but are greatly limited by donor shortage and the need for lifelong immunosuppression. Hence, there is clearly a need for alternative, state-of-the-art treatment options.
Several gene therapy-based strategies have emerged as promising alternatives to current treatments. One potential strategy is to prevent the autoimmune destruction of β cells by delivering genes capable of modifying the immune system or preventing the apoptosis of native β cells [1, 2]. Unfortunately, this strategy relies on the early detection of diabetes, which is difficult given that greater than 80% of an individual’s β cells have often already been destroyed by the time they become symptomatic. Alternatively, researchers have delivered genes encoding β cell-specific transcription factors with the goal of reprogramming non-β cells into β cells capable of synthesizing, storing, and secreting insulin in a glucose-responsive fashion [3-5]. While the liver has been shown, in certain contexts, to have the ability to evade recurring autoimmunity [6], the long-term success of this strategy relies on the persistent survival of newly formed β cells.
To avoid, or at least greatly reduce, the theoretical risk of recurring autoimmunity, researchers have taken a simplified approach termed insulin gene therapy, which aims to merely restore insulin production without duplicating all features of β cells. Insulin gene therapy strategies have targeted a variety of cell types with some success [7], although hepatocytes have emerged as the ideal candidate due to their ability to sense and metabolically respond to fluctuating blood glucose levels. In addition, hepatocytes have a robust capacity for protein synthesis and constitutive secretion. As a result, they have been the most targeted cell type for insulin gene therapy applications. Interestingly, recent work by Ren and colleagues has found that delivery of the preproinsulin gene alone using lentiviral vectors can result in transdifferentiation of hepatocytes into β cell-like cells [8]. While the transdifferentiation of enough cells could theoretically result in near-perfect glucose regulation, the long-term survival and maintenance of these transdifferentiated cells in an environment of preexisting autoimmunity would again become a concern.
To bypass the need to produce surrogate cells resembling β cells, researchers have found alternative strategies to obtain glucose-responsive insulin production. We and others [9-12] have developed insulin expression cassettes containing glucose-inducible response elements (GIREs)-transcriptional enhancer elements that endow glucose-responsive control of gene expression. GIREs generally consist of two E box sequences (CACGTG) separated by five base pairs and serve as the recognition site for two heterodimeric transcription factors: Carbohydrate response element-binding protein and Max-like protein X [13-15]. Our insulin expression cassette 3xGIRE.ALB.Ins1-2xfur contains three copies of GIREs from the S14 gene to endow glucose-responsive transcriptional control of insulin output and the liver-specific albumin promoter to restrict insulin production to hepatocytes-cells with an inherent capacity to sense changing glucose levels. The expression cassette also contains an α-fetoprotein transcriptional enhancer, vascular endothelial growth factor (VEGF) translational enhancer, and albumin 3' UTR to improve insulin output. The preproinsulin coding sequence has been modified to be compatible with furin cleavage to ensure functional maturation of insulin within hepatocytes. We have previously demonstrated that 3xGIRE.ALB.Ins1-2xfur is capable of fully correcting diabetic hyperglycemia in streptozotocin (STZ)-treated diabetic rats [10]. However, the therapeutic benefits only persisted for one month due to the nature of gene delivery vehicles used, adenovirus or minicircle DNA, which are only capable of producing gene expression for a limited period of time.
In this study, we extended the duration of glucose control by delivering 3xGIRE.ALB.Ins1-2xfur using lentiviral vectors gene delivery vehicles capable of stably integrating their genetic cargo into the chromosomes of both dividing and non-dividing cells and thus provide long-term transgene expression. Indeed, we found that delivery of 3xGIRE.ALB.Ins1-2xfur using lentivirus was effective at correcting diabetic hyperglycemia for over one year in STZ-induced diabetic rats, although challenges in affordably producing large-scale batches of lentivirus hampered our ability to optimize dosing. In addition, we observed improved glucose clearance upon challenge and healthy liver function in our LV-treated rats.
Materials and methodsTop
Production and titration of lentiviral vectors
Third-generation HIV1-based lentiviral vectors were produced in 10-layer Nunc Cell Factories (Thermo Scientific; Madison, WI). 293T cells were transfected with 200 µg of the packaging plasmid pMDLg/pRRE, the envelope plasmid pMD.G, and pRSV-Rev and 228 µg pLV-3xGIRE-ALB.rIns1-2xfur by polyethyleneimine (PEI) precipitation. After 17 h, 4 mM caffeine was added to the culture medium [16]. Four days post-transfection, virus-containing culture medium was harvested, filtered, and PEG-precipitated. Following precipitation, virus was collected by centrifugation at 8,700 g for 20 min [17]. Virus pellets were then resuspended in ice-cold PBS, transferred to ultracentrifuge tubes, underlain with 20% sucrose, and centrifuged at 18,000 rpm using a Beckman Coulter SW 28 rotor for two hours at 4°C. Following ultracentrifugation, virus pellets were resuspended in roughly 3 mL ice-cold PBS and flash frozen in liquid nitrogen. Alternatively, LV.3xGIRE.ALB.Ins1-2xfur was produced by American Gene Technologies International, Inc. (Rockville, MD). Lentiviral titers were determined by transducing 293T cells with serially diluted vector stocks, isolating their genomic DNA and treating with RNase A, and quantifying viral integrations using qPCR with the primers found in Table 1. Titers were calculated for each dilution using 2-(Ct,target-Ct,housekeeping), multiplied by a factor of two to normalize for the housekeeping gene, Ubiquitin C (Ubc), being present in two copies per genome, and divided by the volume of lentivirus used per dilution [18].
| Gene | Accession | Primer sequence |
| Primers to determine numbers of lentiviral integrations and titers | ||
| LVψ | F 5'-ATA TAG TAT GGG CAA GCA GGG-3' | |
| R 5'-TGT CTG AAG GGA TGG TTG TAG-3' | ||
| rUbc | NC_005111.4 | F 5'-AGA ACT GTG GGT ATT AGA CGG-3' |
| R 5'-ATC GCA CTC CTG TAT TTA GCA-3' | ||
| hUBC | NG_027722.2 | F 5'-CCA GTC AGT GAC AAA CTT CAC-3' |
| R 5'-GCT CTT ATT CGG GTG AGA TGG-3' | ||
| mUbc | NC_000071.6 | F 5'-ATT CAC AGG TCA GGA ATG ACA-3' |
| R 5'-TGC CGA GGT TTC TTA ACT TTG-3' | ||
| Primers to assess hepatic transdifferentiation | ||
| rlns1 | NM_019129.3 | F 5'-ATC TTC AGA CCT TGG CAC T-3' |
| R 5'-TAG TTC TCC AGT TGG TAG AGG-3' | ||
| rGcg | NM_012707.2 | F 5'-TGT GAG TTC TTA CTT GGA GGG-3' |
| R 5'-ATC AGC CAG TTG ATG AAG TCT-3' | ||
| rSst | NM_012659.2 | F 5'-TCT GGA AGA CAT TCA CAT CCT-3' |
| R 5'-TTC TAA CAC AGG GTC TAG TTG-3' | ||
| rPpy | NM_012626.2 | F 5'-CTC CCT GTT TCT CCT ATC CAC-3' |
| R 5'-TTT CGT ATT GAG CCC TCT GTT-3' | ||
| rPdx1 | NM_022852.3 | F 5'-AGG AGA ATA AGA GGA CCC GTA-3' |
| R 5'-CTC AGT CAA GTT GAG CAT CAC-3' | ||
| rMafA | XM_006241903.2 | F 5'-CTC GCT CAT TTG CTC TGA AG-3' |
| R 5'-TAA GTT CCT CGG CTT CCT CTA-3' | ||
| rUbc | NM_017314.1 | F 5'-GTC AAA CAG GAA GAT ACT CGT-3' |
| R 5'-ATC AAA CCC AAG AAC AAG CAC-3' | ||
| Primers to quantify in vitro glucose induction | ||
| rlns1 | NM_019129.3 | F 5'-ATC TTC AGA CCT TGG CAC T-3' |
| R 5'-TAG TTC TCC AGT TGG TAG AGG-3' | ||
| mUbc | NM_019639.4 | F 5'-TGT TAC CAC CAA GAA GGT CAA-3' |
| R 5'-CAT CAC ACC CAA GAA CAA GC-3' | ||
Lentiviral treatment of STZ-induced diabetic rats
Male Wistar rats weighing 60 to 70 g were rendered diabetic through intraperitoneal injection of 100 mg/kg STZ. Rats that showed fasting blood glucose levels greater than 500 mg/dL on two consecutive days post-STZ treatment were considered diabetic and subsequently used for injection of lentivirus. Diabetic rats were anesthetized using isoflurane and injected with the indicated doses of lentivirus via tail vein injection. Periodic fasting and ad libitum fed blood glucose levels, as well as body weights, of all the rats were measured.
All animal studies were conducted under animal protocols approved by the School of Medicine and Public Health Animal Care and Use Committee (IACUC) at the University of Wisconsin at Madison. Experimental animals were not subjected to any procedures that may cause prolonged pain and were humanely euthanized under anesthesia at the end of the experiments during the process of final blood and tissue sample collection. In accordance with PHS policy on humane care and use of laboratory animals of the NIH, rats becoming sick or accidentally injured were euthanized by hypoxia using CO2. Rodents were placed in a non-precharged chamber and 100% CO2 was introduced at a rate of 10-20% of the chamber volume per minute; rats were kept in the chamber until clinically dead.
Glucose tolerance test
Intraperitoneal glucose tolerance tests (IPGTT) were performed on rats after six hours fast. Blood was drawn to establish a baseline for glucose and insulin levels. An aliquot of 40% glucose solution was then intraperitoneally injected into each test rat to achieve a glucose dose of 3.4 g/kg. Blood glucose measurements were made at the indicated time points over the course of the next three hours. Blood samples were also collected to measure insulin levels. Insulin levels were measured using the Ultrasensitive Rat Insulin ELISA reagents from Mercodia, as described in [10].
Immunohistochemistry
Liver, pancreas, spleen, kidney, and muscle tissues from normal rats, STZ-induced diabetic rats, and STZ-induced diabetic rats treated with various batches of LV.3xGIRE.ALB.Ins1-2xfur were excised at the conclusion of the study, and a portion of the organs were immediately fixed in phosphate buffered 10% formalin (containing 3.7%-4% formaldehyde). After overnight fixation, tissue samples were dehydrated in graded ethanol, infiltrated with xylene, and paraffin-embedded. Paraffin blocks were then cut into 8 μm thick sections, deparaffinized, and rehydrated before immunohistochemical detection of insulin using guinea pig anti-porcine insulin primary antibody, which cross-reacts with rat insulin. A peroxidase-conjugated rabbit anti-guinea pig IgG secondary antibody (Sigma; St. Louis, MO) and Chromogenic ImmPACT DAB peroxidase substrate (Vector Laboratories; Burlingame, CA) were used to visualize insulin localization according to the manufacturer’s procedure. Insulin-stained liver and pancreas sections were lightly stained with hemotoxylin to visualize nuclei. Liver sections from each animal were also stained with hemotoxylin and eosin for analysis of T-cell infiltration.
Nucleic acid extraction and PCR analysis
At the conclusion of the study, liver, pancreas, kidney, spleen, and muscle tissues were harvested and snap-frozen in liquid nitrogen. The tissues were subsequently powdered with a liquid nitrogen-pre-chilled mortar and pestle and immediately transferred into lysis buffer (IBI Scientific; Peosta, IA) containing β-mercaptoethanol. Genomic DNA and total RNA were subsequently isolated using the IBI Scientific DNA/RNA/Protein Extraction Kit following the manufacturer’s instructions. Total RNA was treated with DNase I (TURBO DNA-free; Thermo Scientific), while gDNA was treated with RNase A followed by phenol: Chloroform extraction and ethanol precipitation. Genomic DNA and RNA yields were determined based on A260 using a NanoDrop 1000 Spectrophotometer (Thermo Scientific).
To determine the number of lentiviral integrations per cell, gDNA was diluted to 1 ng/μL and used directly for qPCR analysis on a BioRad C1000 Thermal Cycler with B-R SYBR Green Supermix for iQ (Quanta Biosciences; Beverly, MA). Integrations per cell were calculated in the various tissues using 2-(Ct,target-Ct,housekeeping) and multiplied by a factor of two to normalize for Ubc being present in two copies per genome. To identify hepatic transdifferentiation, 2 μg total RNA isolated from pancreas and liver were reverse transcribed into cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems; Foster City, CA) on an Eppendorf Thermal Cycler according to recommended method of the supplier. Markers of hepatic transdifferentiation into β cells were detected by amplification of 100 ng cDNA using Eppendorf Thermal Cycler and Platinum Taq DNA polymerase (Invitrogen; Carlsbad, CA) with the oligonucleotide primers listed in Table 1. All primer annealing temperatures were optimized empirically, and Ubc expression was used as an internal control. The PCR products were resolved by agarose gel electrophoresis and visualized by ethidium bromide staining.
Ultrastructure analysis by transmission electron microscopy
Pancreas and liver tissues were harvested from rats and immediately placed into Carson Millonig’s fixative (American MasterTech Scientific, Inc.; Lodi, CA), where it was diced into 1 mm cubes. Tissue was fixed for two hours, rinsed with 0.1 M sodium phosphate buffer, dehydrated in a graded series of ethanol, and infiltrated with LR White resin on a rotary shaker overnight. Tissue was transferred to gelatin capsules and solidified at 50°C for 18 h. Tissue was subsequently sectioned with a microtome into 70 nm thick sections and placed on 300 mesh nickel grids (Electron Microscopy Sciences; Hatfield, PA). Sections were rinsed, blocked with 0.5% BSA-C Block (Aurion; Wageningen, Netherlands) for 30 min, incubated with primary and secondary antibodies, and counterstained with 7.7% uranyl acetate. All rinse solutions were filtered through a 0.22 μm SFCA syringe filter immediately before use. The following antibodies were used for detection of insulin and chromogranin A: Mouse anti-chromogranin A (Enzo; Farmingdale, NY), guinea pig anti-insulin (Linco; St. Charles, MO), goat anti-mouse IgG Fc preabsorbed with 18 nm gold (abcam; Cambridge, UK), and donkey anti-guinea pig IgG H&L preabsorbed with 6 nm gold (abcam). Stained sections were visualized on a transmission electron microscope.
Statistical analysis
All quantifiable results were reported as mean values ± standard deviation. Statistical analyses were performed using ANOVA followed by a student’s 2-tailed t-test for correlations using Microsoft Excel. P values < 0.05 were considered significant.
ResultsTop
Lentivirus production
Early efforts to produce lentiviral vectors harboring our glucose-responsive insulin expression cassette (LV.3xGIRE.ALB.Ins1-2xfur) resulted in low infectious titers and undetectable insulin secretion in vitro. As a result, lentivirus production was optimized. To do so, lentivirus harboring the GFP gene (LV.CMV.GFP) was produced and titrated over a 1,000-fold range of dilutions in 293T cells. GFP+ cells were counted to obtain an infectious titer. We found that addition of 3-5 μM caffeine 17 h post-transfection led to ∼3-fold increase in infectious titers (data not shown). Optimization of plasmid ratios resulted in a 2.5-fold improvement in titer, while collection of lentivirus four days after transfection led to another 7-fold increase in titer compared to two days after transfection (data not shown). Finally, scaling up production to 10-layer Cell Factories and concentrating the lentivirus by ultracentrifugation over a sucrose cushion led to a 2.5 and 70-fold increase in infectious titer, respectively (data not shown). Altogether, infectious titers showed ∼9,000-fold improvement compared to early batches of unconcentrated virus, ultimately reaching titers greater than 1 × 1010 IFU/mL.
Glucose-stimulated insulin production in vitro
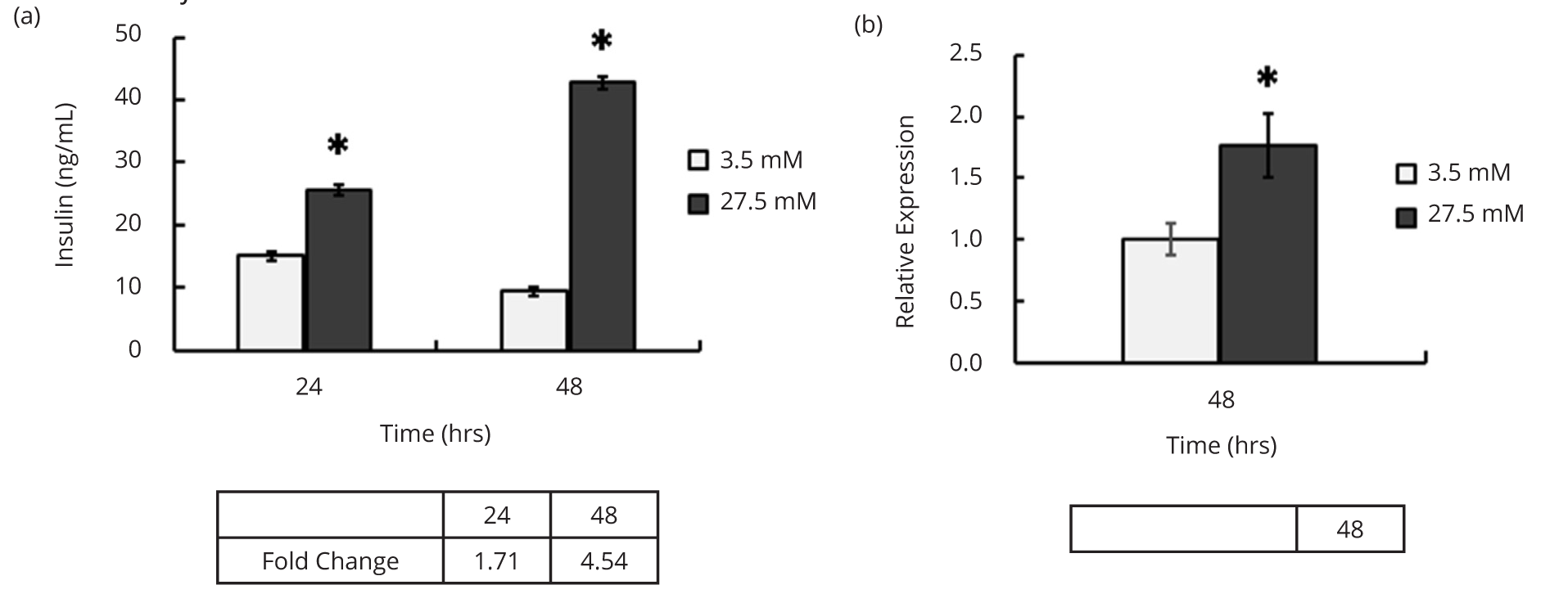
The optimized procedure to generate high titer LV.CMV.GFP was used to produce LV.3xGIRE.ALB.Ins1-2xfur with titers ranging 1-3 x 1010 IFU/mL. Importantly, the improved titers were sufficient to allow detection of insulin in the media from LV.3xGIRE.ALB.Ins1-2xfur-transduced Hepa1-6 cells a mouse hepatoma cell line capable of responding to varying glucose concentrations. To confirm the glucose-responsive insulin production by LV.3xGIRE.ALB.Ins1-2xfur, Hepa1-6 cells were transduced with LV.3xGIRE.ALB.Ins1-2xfur and incubated in media containing either 3.5 mM (low) or 27.5 mM (high) glucose for two days. The media was then collected after 24 and 48 h and analyzed for insulin content by ELISA. RNA was also isolated after 48 h to confirm glucose induction. We found that LV.3xGIRE.ALB.Ins1-2xfur induced a significant 1.7 and 4.5-fold increase in insulin production in high glucose conditions compared to low glucose after 24 and 48 h, respectively (p < 0.0001 at both time points) (Figure 1a). Similarly, LV.3xGIRE.ALB.Ins1-2xfur led to a significant 1.7-fold increase in rIns1 mRNA after 48 h (p = 0.01) (Figure 1b). These findings demonstrate that 3xGIRE.ALB.Ins1-2xfur remains glucose-responsive when delivered via lentivirus.
Treatment of STZ-induced diabetic rats with LV.3xGIRE.ALB.Ins1-2xfur
We next determined whether LV.3xGIRE.ALB.Ins1-2xfur was capable of correcting hyperglycemia in STZ-induced diabetic rats. To do so, we injected diabetic rats with doses of LV.3xGIRE.ALB.Ins1-2xfur ranging from 1.2 to 6.8 x 1011 IFU/kg rat by tail vein injection and measured both ad libitum fed and fasted blood glucose levels. Four days post-lentivirus treatment, blood glucose levels began to improve. We found that a dose of 1.2 x 1011 IFU/kg dubbed “low dose”-led to an insignificant change in blood glucose levels (mean ad libitum fed blood glucose = 588 ± 36 mg/dL; mean fasted blood glucose = 541 ± 64 mg/dL) compared to untreated diabetic control rats (mean ad libitum fed blood glucose = 581 ± 46 mg/dL; mean fasted blood glucose = 556 ± 36 mg/dL) (Figures 2b and 2c). Doses ranging from 2.0 to 2.2 x 1011 IFU/kg—dubbed “medium dose”-induced a partial correction in both ad libitum fed (525 ± 55 and 479 ± 68 mg/dL, respectively) and fasted blood glucose levels (320 ± 161 and 325 ± 132 mg/dL, respectively), while doses ranging from 3.4 to 6.8 × 1011 IFU/kg-termed “high dose”-led to hypoglycemia in rats when fed ad libitum (46 ± 36 and 44 ± 27 mg/dL, respectively); hypoglycemic rats were not fasted and were maintained on sugar water (4-12%). Reduced blood glucose levels persisted for the duration of the study-482 days (Figure 2a).
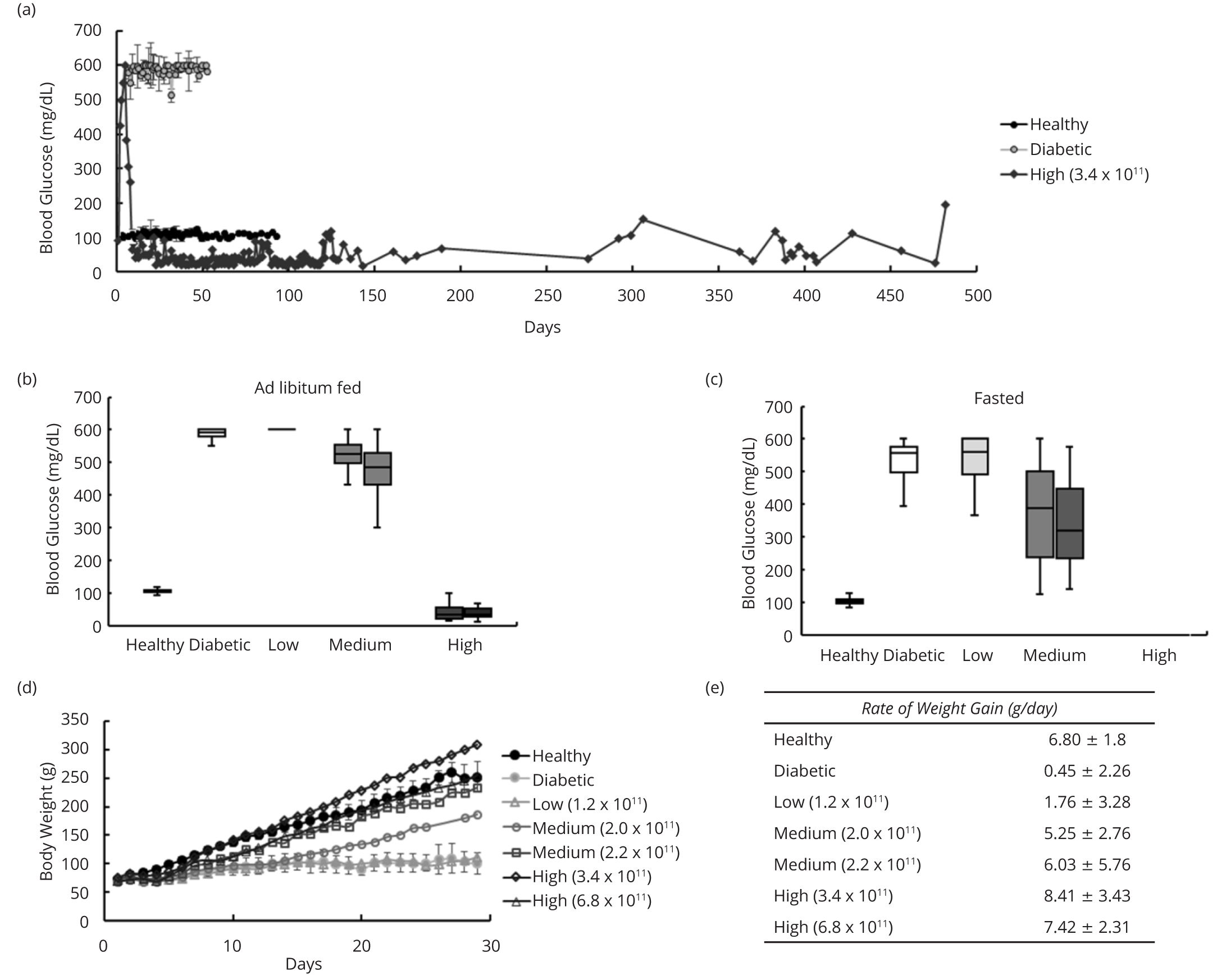
Similarly, STZ-induced diabetic rats treated with LV.3xGIRE.ALB.Ins1-2xfur showed a dose-dependent improvement in their rate of weight gain over the first 30 days post-treatment. Diabetic rats receiving the low dose of LV.3xGIRE.ALB.Ins1-2xfur showed a rate of weight gain of 1.8 ± 3.3 g/day, marginally improved compared to diabetic control rats (0.4 ± 2.3 g/day) (Figures 2d, e). Diabetic rats receiving the medium dose showed rates of weight gain of 5.3 ± 2.8 and 6.0 ± 5.8 g/day, similar to healthy control rats (6.8 ± 1.8 g/day). Rats receiving the high dose of LV.3xGIRE.ALB.Ins1-2xfur tended to gain weight at a quicker rate than healthy control rats, showing rates of 8.4 ± 3.4 and 7.4 ± 2.3 g/day.
Intraperitoneal glucose tolerance test
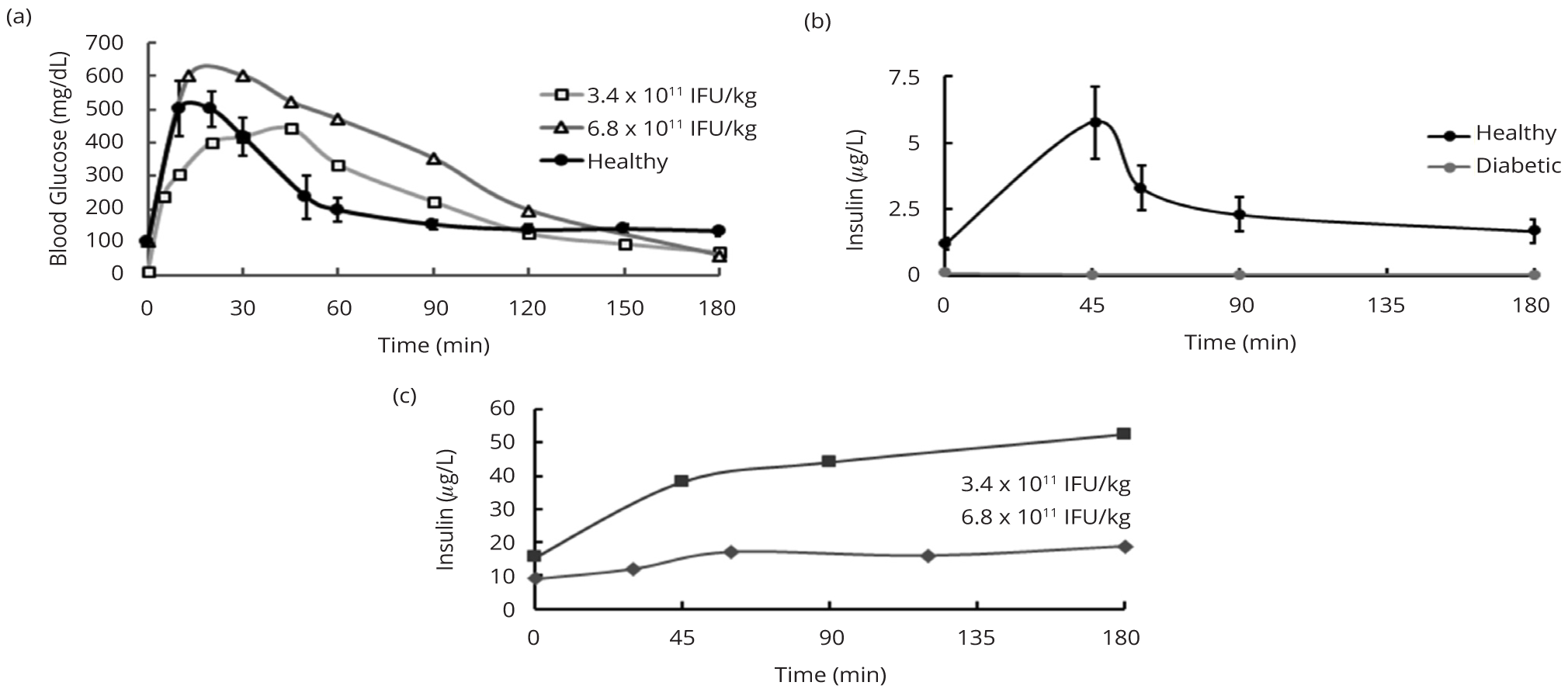
In order to test the ability of rats treated with LV.3xGIRE.ALB.Ins1-2xfur to dispose of a substantial glucose load, an IPGTT was employed using 3.4 g glucose/kg body weight. In healthy control rats, glucose levels peaked around 10 to 20 min after glucose challenge, after which they began to come down (Figure 3a). Glucose levels were restored to relatively normal levels around 90 min after glucose challenge. LV-treated diabetic rats receiving the high dose showed delayed rates of glucose clearance. The rat receiving 6.8 × 1011 IFU/kg displayed a peak in glucose levels 15 to 30 min after challenge before showing steady clearance. The rat receiving 3.4 × 1011 IFU/kg showed a peak in glucose levels around 45 min before steadily declining. In each case, glucose levels returned to normal around 120 to 150 min post-challenge.
Blood was collected at various time points throughout the IPGTT to measure plasma insulin levels via ELISA. We found that healthy control rats showed a nearly 5-fold peak in insulin levels by 45 min post-challenge (4.8 ± 0.6 μg/L), after which insulin was quickly cleared to near normal levels (0.46 ± 0.13 μg/L) by 90 minutes post-challenge (Figure 3b). High dose LV-treated diabetic rats showed at least 5-fold higher basal insulin levels than healthy control rats but a more modest 1.7-fold increase in insulin levels by 45 min post-challenge that gradually increased through three hours post-challenge, demonstrating in vivo glucose induction of insulin production but deficient clearance of insulin (Figure 3c). Untreated diabetic control rats showed negligible insulin levels throughout the IPGTT (Figure 3b).
Lentiviral integration and insulin production in vivo
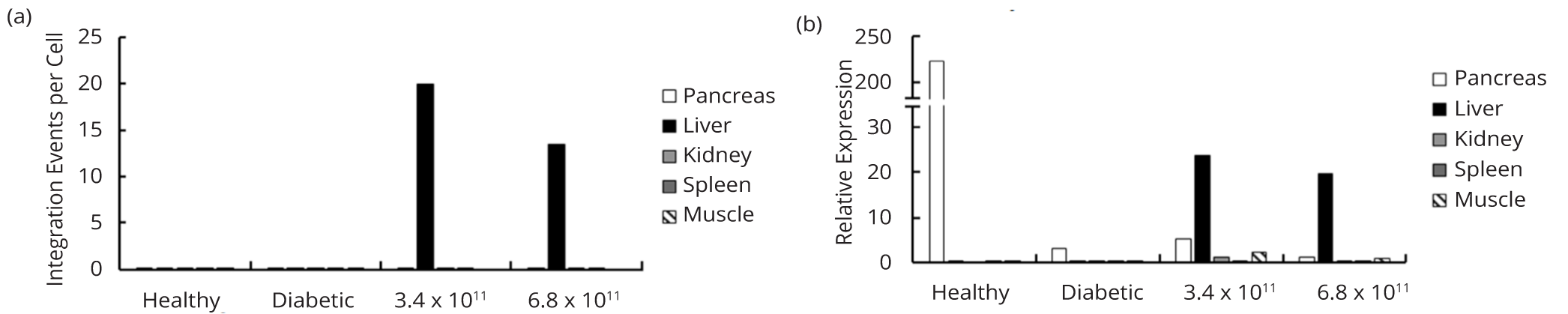
To evaluate lentiviral integration in various tissues in vivo, gDNA was isolated from the pancreas, liver, kidney, spleen, and muscle of rats receiving the high doses of lentivirus, and the number of lentiviral integrations per cell was determined using qPCR with primers directed at the ψ packaging signal. Rats receiving the high dose of lentivirus showed 20.0 and 13.5 lentiviral integrations per cell in the liver (Figure 4a). In each LV-treated rat, we were unable to detect any lentiviral integrations in the pancreas, kidney, or muscle. We did, however, detect 0.13 and 0.16 lentiviral integrations per cell in the spleen of rats receiving the high doses of lentivirus. Nevertheless, negligible splenic expression of rIns1 was detected (Figure 4b), thus validating the liver specificity of insulin expression. Expression of rIns1 was readily detected in the livers of LV-treated rats but remained 9.4 to 11.4-fold lower than in the pancreas of healthy control rats (Figure 4b). While qPCR showed residual expression of rIns1 in the pancreata of both LV-treated and untreated diabetic rats, endogenous rIns1 mRNA levels were reduced between 43 and 175-fold compared to healthy control rats (Figure 4b).
In addition, we found abundant insulin immunostaining in the livers of rats receiving the high doses of LV.3xGIRE.ALB.Ins1-2xfur. Staining was found throughout the liver but tended to be more concentrated in perivascular hepatocytes (Figures 5a and 5b). The pancreata of LV-treated diabetic rats showed islets devoid of insulin immunostaining (Figure 5c), although residual insulin immunostaining was present in some islets (Figure 5d). No staining was found in the livers of untreated diabetic control rats (Figure 5e). As expected, insulin-positive islets were abundant in the pancreata of healthy control rats (Figure 5f).
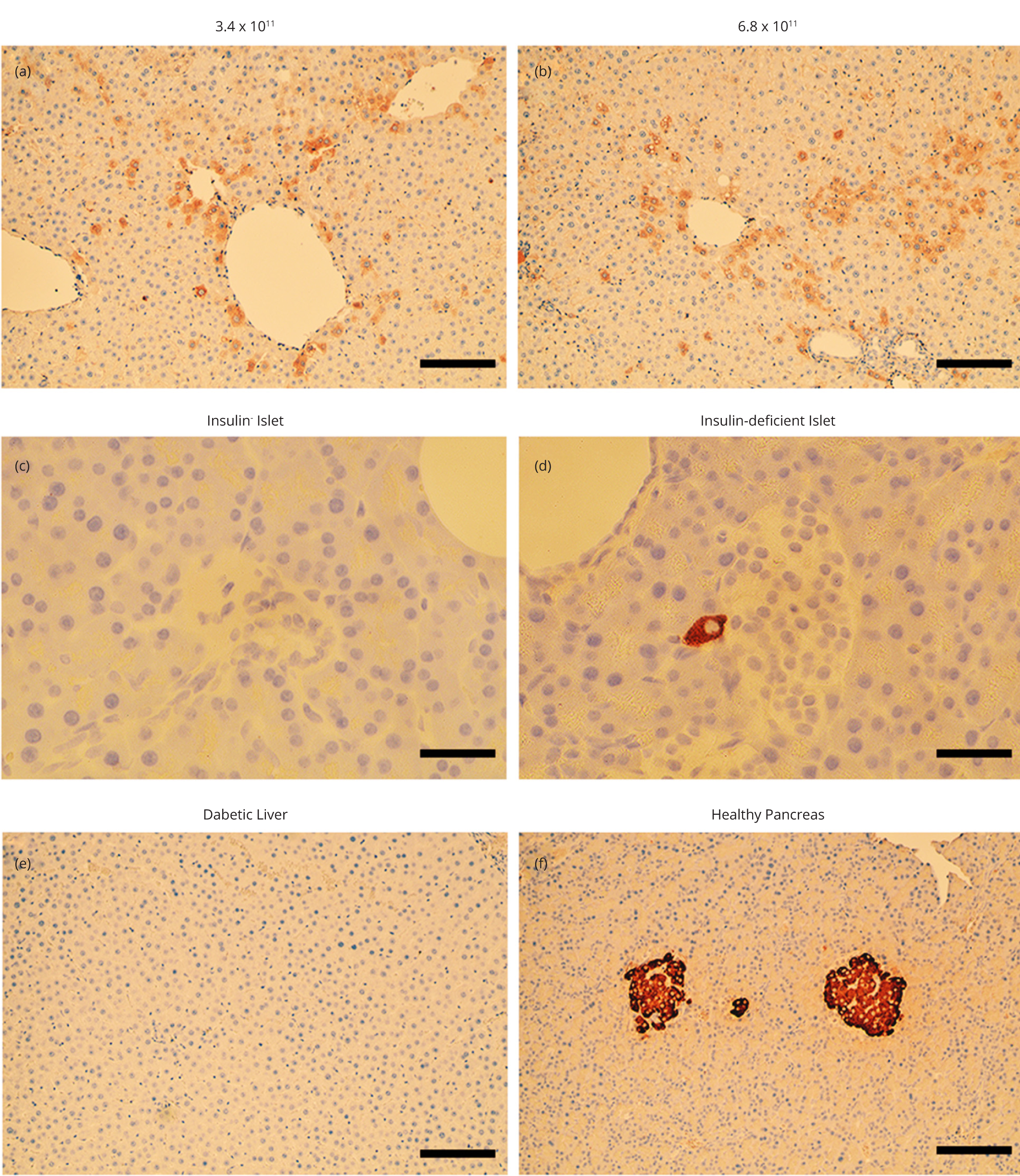
Liver health
Persistent hyperglycemia causes serious metabolic dysregulation, including impaired liver function, hyperlipidemia, hypercholesterolemia, and ketosis. End-point blood tests from healthy rats and LV-treated rats showed restoration of various diabetes-associated markers of metabolic dysregulation and liver function to near normal levels (Table 2). End-point blood test results could not be attained for age-matched diabetic control rats due to declining health, but previously published data from younger STZ-induced diabetic rats showed dramatically elevated levels of aspartate transaminase, alanine aminotransferase, alkaline phosphatase, and cholesterol [10].
| Healthy controls | 3.4 × 1011 IFU/kg | 6.8 × 1011 IFU/kg | |
| Aspartate transaminase (U/L) | 147 ± 18 | 210 | 273 |
| Alanine aminotransferase (U/L) | 77 ± 4 | 26 | 91 |
| Alkaline phosphatase (U/L) | 84 ± 20 | 57 | 72 |
| Albumin (g/dl) | 2.4 ± 0.1 | 2 | 3 |
| Globulin (g/dl) | 2.9 ± 0.1 | 3 | 3 |
| Urea N (mg/dl) | 17 ± 4 | 16 | 22 |
| Cholesterol (mg/dl) | 73 ± 13 | 82 | 68 |
| HDL (mg/dl) | 22 ± 7 | 19 | 31 |
| LDL (mg/dl) | 6.7 ± 0.1 | 11 | 7 |
Note: End-point blood samples were collected from healthy control (n = 3) and high dose LV-treated diabetic rats for serum lipid profile and measurements of liver damage and function. LV-treated rats showed healthy liver function. The values for healthy control rats are presented as mean ± standard deviation.
Liver-to-endocrine pancreas transdifferentiation
To determine if transdifferentiation of hepatocytes occurred in LV-treated rats, we assessed the hepatic expression of several islet hormones and β cell transcription factors in rats injected with the high doses of LV.3xGIRE.ALB.Ins1-2xfur using RT-PCR. We found no detectable expression of the islet-specific transcripts-glucagon (Gcg), somatostatin (Sst), or pancreatic polypeptide (Ppy)-in the livers of LV-treated rats despite observing abundant expression of rIns1 (Figure 6a). While low levels of expression of the β cell transcription factors-pancreatic and duodenal homeobox 1 (Pdx1) and MAF bZIP transcription factor A (Mafa)—were observed in one of the high dose rats, we observed a similar expression pattern in our untreated diabetic control rats. Similar to the qPCR data in Figure 4b, there remained a faint but markedly reduced signal for rIns1 in the pancreata of all three STZ-treated rats. The expression of all other markers was still detected in the pancreata of normal, diabetic, and LV-treated rats at fairly consistent levels. In addition, TEM images of immunogold-labeled sections showed no evidence of the formation of large insulin secretory granules within the livers of LV-treated rats (Figure 6b). In contrast, the pancreata of healthy control rats showed well-defined secretory granules with abundant immunogold-labeled insulin. Together, these data demonstrate no apparent transdifferentiation of hepatocytes into β cells in our LV-treated rats.
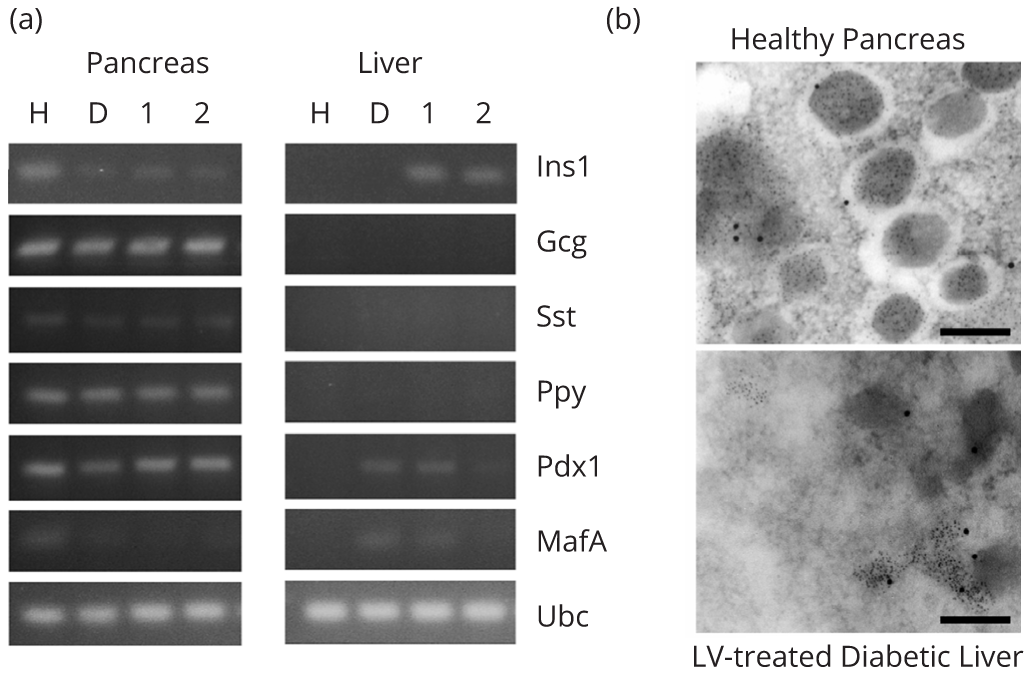
DiscussionTop
For insulin gene therapy to serve as a successful treatment for T1DM, the strategy should ideally (i) target a glucose-sensitive organ, (ii) express insulin in a glucose-responsive fashion, and (iii) sustain long-term insulin expression. Our previous work demonstrated that 3xGIRE.ALB.Ins1-2xfur (formerly called TA1) our GIRE-containing insulin expression cassette is capable of providing both liver specificity and glucose-inducible control of insulin expression [10]. However, these studies delivered 3xGIRE.ALB.Ins1-2xfur using adenovirus or DNA minicircles, thus providing only short-term correction of hyperglycemia. In this study, we extended upon these findings to show that delivery of 3xGIRE.ALB.Ins1-2xfur using lentivirus can provide a long-term correction of diabetic hyperglycemia that is both hepatocyte-specific and glucose-responsive while maintaining healthy liver function. Specifically, we found a correction of diabetes for 482 days (the end-point of the study) (Figure 2a). Insulin expression was specifically targeted to the liver, as evidenced most notably by qPCR results showing 13 to 20 lentiviral integrations in the liver of our high dose rats compared to negligible integration in their pancreas, kidney, spleen, or muscle (Figure 4a). Further, glucose induction of insulin expression was evident from the 4.5-fold increase in insulin secretion in vitro in high glucose conditions (Figure 1a) and 1.7-fold increase in serum insulin levels during the IPGTT (Figure 3c).
Other groups have likewise found that lentiviral vectors are capable of providing long-term correction of diabetic hyperglycemia in rodent models. For instance, Elsner and colleagues delivered furin-cleavable insulin to the livers of autoimmune diabetic and STZ-induced diabetic rats via portal vein injection and found restoration of ad libitum fed blood glucose levels within a normal range for one year [19]. Elsner and colleagues injected 7 × 109 lentiviral particles per rat in their highest dose. Interestingly, we found no improvement in diabetic hyperglycemia after injecting 9.2 × 109 lentiviral particles per rat (Figures 2b and 2c; low dose), although the weights of their rats may have been drastically different at the time of injection. Similarly, Ren and coworkers delivered furin-cleavable insulin to the livers of non-obese diabetic mice using lentivirus through intervallic infusion during full flow occlusion and found correction of diabetic hyperglycemia for at least 150 days [8]. Using their surgical technique to transiently restrict the hepatic blood flow, restoration of normal blood glucose levels only required 8 × 108 infectious lentiviral particles. Interestingly, the optimal doses used in both of these studies were likely substantially lower than our anticipated optimal dose of around 2.8 × 1011 IFU/kg. This is expected given that both groups delivered the preproinsulin gene under the control of strong, constitutive promoters (cytomegalovirus and HIV/murine stem cell virus hybrid long terminal repeat promoters, respectively). However, use of these promoters entails an absence of glucose-responsive control of insulin expression; notably, neither study reported fasting blood glucose levels. In addition to differences in promoter, Ren and colleagues delivered the lentiviral vectors by direct injection during full flow occlusion and also observed hepatic transdifferentiation, both of which could further lower the dose of lentiviral vectors necessary for correction of hyperglycemia. To our knowledge, we are the first group to use lentivirus to deliver the preproinsulin gene under the control of a glucose-responsive and tissue-specific promoter.
Challenges with large-scale production of lentiviral vectors hindered our ability to optimize dose and establish glucose levels within a normal range in diabetic animals. As a result, two of the rats showed a partial correction of hyperglycemia, while another two rats became hypoglycemic. To improve lentivirus titer, we made several modifications but remained unable to treat more than one rat with a single batch of lentivirus. These modifications included (i) optimization of the lentivirus plasmid ratio for co-transfection in 293T cells, (ii) addition of caffeine to the media 17 h post-transfection [16], and (iii) collection of virus four days after transfection instead of two or three days. Treatment of diabetes by insulin gene therapy requires precise dosing of the preproinsulin gene to bring glucose levels within a normal range without causing hypoglycemia. This is in contrast to many other gene therapy applications, which only require a sustained level of therapeutic gene without dynamic control. Simpler yet, other applications like hemophilia require only a partial restoration in clotting factor levels to achieve an improvement in disease phenotype [20]. Thus, further improvements to lentivirus production will be critical if gene therapy is to be used for treatment of T1DM.
An alternative strategy to lessen the burden of large-scale production would be to make modifications to the lentiviral particles to improve their in vivo infectivity. One potential improvement would be to engineer VSV-G envelope proteins with enhanced functionality. For example, Hwang and Schaffer used directed evolution to identify VSV-G mutants with increased resistance to human serum inactivation [21]. After multiple selection cycles, they found several common mutations to surface exposed residues that not only increased serum resistance but also enhanced thermostability. Ultimately, the VSV-G mutants showed a nearly 3-fold improvement in transduction efficiency in vivo in the presence of human serum. Similarly, Kaikkonen and colleagues generated a truncated VSV-G mutant retaining only the transmembrane and cytoplasmic tail domains of VSV-G [22]. While this study was performed in a baculovirus model, they showed a nearly 15-fold increase in transduction efficiency compared to control virus, demonstrating its potential utility in improving lentivirus-based gene transfer. These VSV-G mutants, along with other as yet unidentified mutants, could ameliorate current issues related to high therapeutic dose and the necessity for large-scale lentivirus production.
In this study, two observations strongly suggest that further modifications to the insulin expression cassette 3xGIRE.ALB.Ins1-2xfur may be necessary in order to attain adequate glucose regulation: (i) The dose range required to restore blood glucose levels within a normal range is very narrow. A dose of 2.2 × 1011 IFU/kg resulted in a partial correction of diabetic hyperglycemia, whereas a dose of 3.4 × 1011 IFU/kg produced hypoglycemic rats that needed to be maintained on sugar water. Given the inherent differences in immune systems from one person to another, this limitation could severely complicate any efforts to translate this research to a clinical setting; (ii) Insulin ELISA data from the IPGTT showed a modest 1.75-fold increase in serum insulin levels upon challenge that continued to rise through three hours post-challenge. By this time, glucose levels had already been restored to baseline levels, raising concerns of a hypoglycemic overshoot. Together, these observations strongly suggest a need for further improvements to 3xGIRE.ALB.Ins1-2xfur. Namely, it would be ideal to increase the degree of glucose-induced insulin transcription [23] while also incorporating mechanism(s) to more quickly reduce insulin levels upon restoration of normoglycemia [24]. Overall, such modifications would greatly enhance the dynamic insulin response to changes in glucose levels, leading to a more effective and safer strategy with greater latitude in dosing.
In our work, we found no evidence of pancreatic transdifferentiation within the livers of LV-treated rats. This is in contrast with the work from Ren and colleagues, who observed the hepatic expression of a variety of β cell transcription factors, as well as murine insulin, glucagon, and somatostatin. They also observed signs of insulin in hepatic secretory granules [8]. As the authors noted, the observed transdifferentiation in their studies was likely caused by the surgical technique used, which isolated the liver from its blood supply during injection and caused transient ischemia. Interestingly, transient hypoxia has been found to be a critical environmental cue for the acquisition and maintenance of stem cell-like qualities [25]. For instance, mesenchymal stem cells (MSCs) tend to be found throughout the body in areas of low oxygen tension [26, 27], and when cultured ex vivo, hypoxic conditions have been shown to extend their replicative lifespan [28, 29], maintain their multipotent differentiation potential into later passages [30, 31], and upregulate the expression of stemness markers [32]. More remarkably, hypoxia has been shown to enhance the reprogramming efficiency of somatic cells into induced pluripotent stem cells (iPSCs) [33, 34]. Thus, their surgical technique may have caused hepatic dedifferentiation followed by subsequent redifferentiation into an islet phenotype once blood flow was restored. If true, given that we delivered lentivirus through tail vein injection, it is not surprising that hepatic transdifferentiation was not observed in this study.
ConclusionTop
Overall, we demonstrated the ability to address all three of the aforementioned criteria to create a long-term, glucose-responsive treatment for T1DM using lentivirus. Moving forward, we must greatly improve our ability to produce large batches of lentivirus affordably so that a single batch can treat more than one rat. We may also need to enhance the infectivity of the lentiviral particles produced. In addition, it would be advantageous to further refine 3xGIRE.ALB.Ins1-2xfur to incorporate more glucose-responsive elements, thus widening the therapeutic dose for effective treatment and greatly reducing the risk of hypoglycemia. Without multiple levels of glucose-responsive regulation, it will be difficult to create a treatment using insulin gene therapy that can maintain blood glucose levels within a safe range under normal physiological states.
Acknowledgments
We thank the Madison Veteran’s Hospital Electron Microscopy Lab for their advice and assistance in preparing and imaging our immunogold-labeled tissues and American Gene Technologies International, Inc. (Rockville, MD) for providing some lentivirus preparations used in this study. We are grateful to Dr. Inder Verma (Salk Institute) for his valuable advice and providing plasmids necessary for lentivirus production.
Funding
This work was supported by intramural funds from the University of Wisconsin-Madison Department of Surgery (grant number 233AT84).
Conflicts of interest
The authors declare no conflicts of interest.
ReferencesTop
[1]Cameron MJ, Arreaza GA, Waldhauser L, Gauldie J, Delovitch TL. Immunotherapy of spontaneous type 1 diabetes in nonobese diabetic mice by systemic interleukin-4 treatment employing adenovirus vector-mediated gene transfer. Gene Ther. 2000; 7(21):1840–1846.Article Pubmed
[2]Allison J, Thomas H, Beck D, Brady JL, Lew AM, et al. Transgenic overexpression of human Bcl-2 in islet beta cells inhibits apoptosis but does not prevent autoimmune destruction. Int Immunol. 2000; 12(1):9–17.Article Pubmed
[3]Ferber S, Halkin A, Cohen H, Ber I, Einav Y, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000; 6(5):568–572.Article Pubmed
[4]Ber I, Shternhall K, Perl S, Ohanuna Z, Goldberg I, et al. Functional, persistent, and extended liver to pancreas transdifferentiation. J Biol Chem. 2003; 278(34):31950–31957.Article Pubmed
[5]Shternhall-Ron K, Quintana FJ, Perl S, Meivar-Levy I, Barshack I, et al. Ectopic PDX-1 expression in liver ameliorates type 1 diabetes. J Autoimmun. 2007; 28(2-3):134–142.Article Pubmed
[6]Tabiin MT, White CP, Morahan G, Tuch BE. Insulin expressing hepatocytes not destroyed in transgenic NOD mice. J Autoimmune Dis. 2004; 1(1):3.Article Pubmed
[7]Handorf AM, Sollinger HW, Alam T. Insulin gene therapy for type 1 diabetes mellitus. Exp Clin Transplant. 2015; 13(Suppl 1):37–45.Article Pubmed
[8]Ren BH, O'Brien BA, Byrne MR, Ch'ng E, Gatt PN, et al. Long-term reversal of diabetes in non-obese diabetic mice by liver-directed gene therapy. J Gene Med. 2013; 15(1):28–41.Article Pubmed
[9]Alam T, Sollinger HW. Glucose-regulated insulin production in hepatocytes. Transplantation. 2002; 74(12):1781–1787.Article Pubmed
[10]Alam T, Wai P, Held D, Vakili ST, Forsberg E, et al. Correction of diabetic hyperglycemia and amelioration of metabolic anomalies by minicircle DNA mediated glucose-dependent hepatic insulin production. PLoS One. 2013; 8(6):e67515.Article Pubmed
[11]Thule PM, Liu J, Phillips LS. Glucose regulated production of human insulin in rat hepatocytes. Gene Ther. 2000; 7(3):205–214.Article Pubmed
[12]Han J, McLane B, Kim EH, Yoon JW, Jun HS. Remission of diabetes by insulin gene therapy using a hepatocyte-specific and glucose-responsive synthetic promoter. Mol Ther. 2011; 19(3):470–478.Article Pubmed
[13]Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. 2001; 98(16):9116–9121.Article Pubmed
[14]Stoeckman AK, Ma L, Towle HC. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J Biol Chem. 2004; 279(15):15662–15669.Article Pubmed
[15]Ma L, Tsatsos NG, Towle HC. Direct role of ChREBP center dot Mlx in regulating hepatic glucose-responsive genes. J Biol Chem. 2005; 280(12):12019–12027.Article Pubmed
[16]Ellis BL, Potts PR, Porteus MH. Creating higher titer lentivirus with caffeine. Hum Gene Ther. 2011; 22(1):93–100.Article Pubmed
[17]Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006; 1(1):241–245.Article Pubmed
[18]Geraerts M, Willems S, Baekelandt V, Debyser Z, Gijsbers R. Comparison of lentiviral vector titration methods. BMC Biotechnol. 2006; 6:34.Article Pubmed
[19]Elsner M, Terbish T, Jorns A, Naujok O, Wedekind D, et al. Reversal of diabetes through gene therapy of diabetic rats by hepatic insulin expression via lentiviral transduction. Mol Ther. 2012; 20(5):918–926.Article Pubmed
[20]Cantore A, Ranzani M, Bartholomae CC, Volpin M, Valle PD, et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med. 2015; 7(277):277ra228.Article Pubmed
[21]Hwang BY, Schaffer DV. Engineering a serum-resistant and thermostable vesicular stomatitis virus G glycoprotein for pseudotyping retroviral and lentiviral vectors. Gene Ther. 2013; 20(8):807–815.Article Pubmed
[22]Kaikkonen MU, Raty JK, Airenne KJ, Wirth T, Heikura T, et al. Truncated vesicular stomatitis virus G protein improves baculovirus transduction efficiency in vitro and in vivo. Gene Ther. 2006; 13(4):304–312.Article Pubmed
[23]Zhang T, Dong HH. Glucose-regulated insulin production in the liver improves glycemic control in type 1 diabetic mice. Mol Metab. 2015; 4(1):70–76.Article Pubmed
[24]Thule PM, Lin Y, Jia D, Olson DE, Tang SC, et al. mRNA destabilization improves glycemic responsiveness of transcriptionally regulated hepatic insulin gene therapy in vitro and in vivo. J Gene Med. 2017; 19(3):70–76.Article
[25]Brown PT, Handorf AM, Jeon WB, Li WJ. Stem cell-based tissue engineering approaches for musculoskeletal regeneration. Curr Pharm Des. 2013; 19(19):3429–3445.Article Pubmed
[26]Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008; 3(3):301–313.Article Pubmed
[27]Shi S, Gronthos S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J Bone Miner Res. 2003; 18(4):696–704.Article Pubmed
[28]Fehrer C, Brunauer R, Laschober G, Unterluggauer H, Reitinger S, et al. Reduced oxygen tension attenuates differentiation capacity of human mesenchymal stem cells and prolongs their lifespan. Aging Cell. 2007; 6(6):745–757.Article Pubmed
[29]Tsai CC, Chen YJ, Yew TL, Chen LL, Wang JY, et al. Hypoxia inhibits senescence and maintains mesenchymal stem cell properties through down-regulation of E2A-p21 by HIF-TWIST. Blood. 2011; 117(2):459–469.Article Pubmed
[30]Adesida AB, Mulet-Sierra A, Jomha NM. Hypoxia mediated isolation and expansion enhances the chondrogenic capacity of bone marrow mesenchymal stromal cells. Stem Cell Res Ther. 2012; 3(2):9.Article Pubmed
[31]Volkmer E, Kallukalam BC, Maertz J, Otto S, Drosse I, et al. Hypoxic preconditioning of human mesenchymal stem cells overcomes hypoxia-induced inhibition of osteogenic differentiation. Tissue Eng Part A. 2010; 16(1):153–164.Article Pubmed
[32]Hung SP, Ho JH, Shih YR, Lo T, Lee OK. Hypoxia promotes proliferation and osteogenic differentiation potentials of human mesenchymal stem cells. J Orthop Res. 2012; 30(2):260–266.Article Pubmed
[33]Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. 2009; 5(3):237–241.Article Pubmed
[34]Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci U S A. 2005; 102(13):4783–4788.Article Pubmed
 Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved