



- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Radiology and Imaging
Volume 1, Issue 2, August 2016, Pages 14–17
Case reportOpen Access
Fetal intracranial neoplasm–not always a teratoma!
-
Leslie E. Hirsig1 and
Dhanashree A. Rajderkar1,*

*Corresponding author: Dhanashree A. Rajderkar, M.D., Department of Radiology, University of Florida - College of Medicine, 1600 SW Archer Rd, P.O. Box 100374, Gainesville, FL 32610-0374, United States. Mobile: 314-71-7990; Fax: 352-265-0279; E-mail: RAJDDA@radiology.ufl.edu
Received 3 May 2016 Revised 29 June 2016 Accepted 15 July 2016 Published 25 July 2016
DOI: http://dx.doi.org/10.14312/2399-8172.2016-4
Copyright: © 2016 Hirsig LE, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
Although congenital intracranial tumors are very rare, it is important to know the differential diagnosis and distinguishing features of the different disease processes in order to accurately diagnosis and appropriately treat these patients in the neonatal period. We present a case of a rare congenital craniopharyngioma detected in a fetus on prenatal imaging. Teratoma is the most common congenital intracranial tumor. Hence this tumor was initially labelled as a teratoma, which is a pitfall that should be avoided.
Keywords: craniopharyngioma; fetal; congenital; intracranial neoplasm
IntroductionTop
Intracranial tumors are rare in the fetal and neonatal period. The most common brain tumor identified congenitally is a teratoma [1]. However, it is important to keep other rare pathologies in mind. The case presented was initially thought to represent a teratoma, but postnatal imaging characteristics disfavored that diagnosis in this biopsy-proven congenital craniopharyngioma.
Clinical presentationTop
A 24-year-old pregnant female underwent a fetal ultrasound at 33 weeks gestation. The fetus was noted to have severe hydrocephalus. Upon further inspection, a 6cm brain tumor was identified in the region of the third ventricle. Of note, the 20 weeks fetal ultrasound was reportedly normal. A fetal MRI was therefore requested to characterize the mass and evaluate for metastatic disease.
The fetal MRI was obtained at 34 weeks gestation. A 6.5 × 5.8 × 5.0 cm heterogeneous, midline suprasellar mass was demonstrated in the third ventricle on T2-weighted images (Figure 1a, 1b). Multiple spontaneously bright T1 foci were also noted suggesting presence of blood products or fat (Figure 2). There was secondary severe obstructive hydrocephalus of the lateral ventricles and marked diffuse thinning of the cortical mantle. Findings on the fetal MRI were favored to represent a teratoma, but the differential diagnosis included craniopharyngioma, germ cell tumor, atypical teratoid rhabdoid tumor and glioma [1, 2]. Given the location and the morphology, craniopharyngioma should to be considered as the top differential.
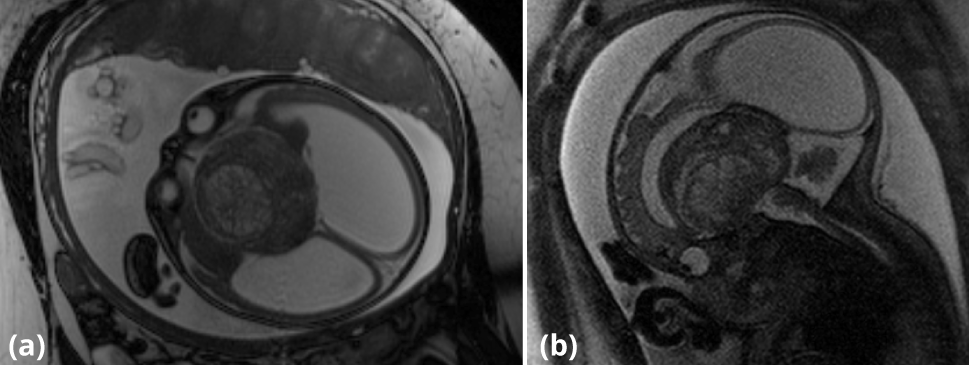
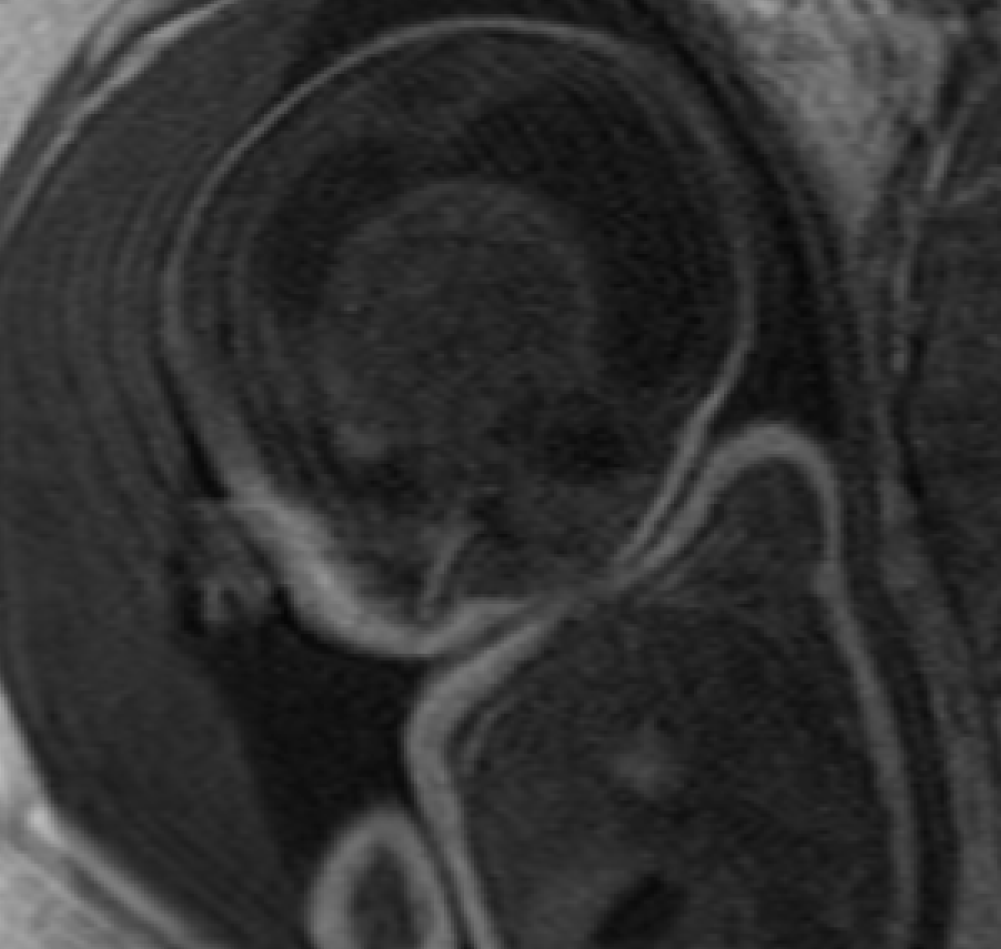
A classical cesarean section was performed at 36 weeks due to pre-term labor. The neonate was in distress at birth requiring intubation and admission to the neonatal intensive care unit.
Postnatal MRI of the brain on day 3 of life again demonstrated severe obstructive hydrocephalus secondary to a large, heterogeneous suprasellar mass, which had increased in size to 7.0 × 5.8 × 5.3cm (Figure 3a - 3e). There was no restricted diffusion. MR spectroscopy was also performed and demonstrated marked suppression of the N-acetylaspartate (NAA) peak with marked elevation of choline as well as increased lactic acid within the central portion of mass (Figure 4). The spectroscopy results therefore suggest a high-grade neoplasm with central necrosis rather than benign teratoma.
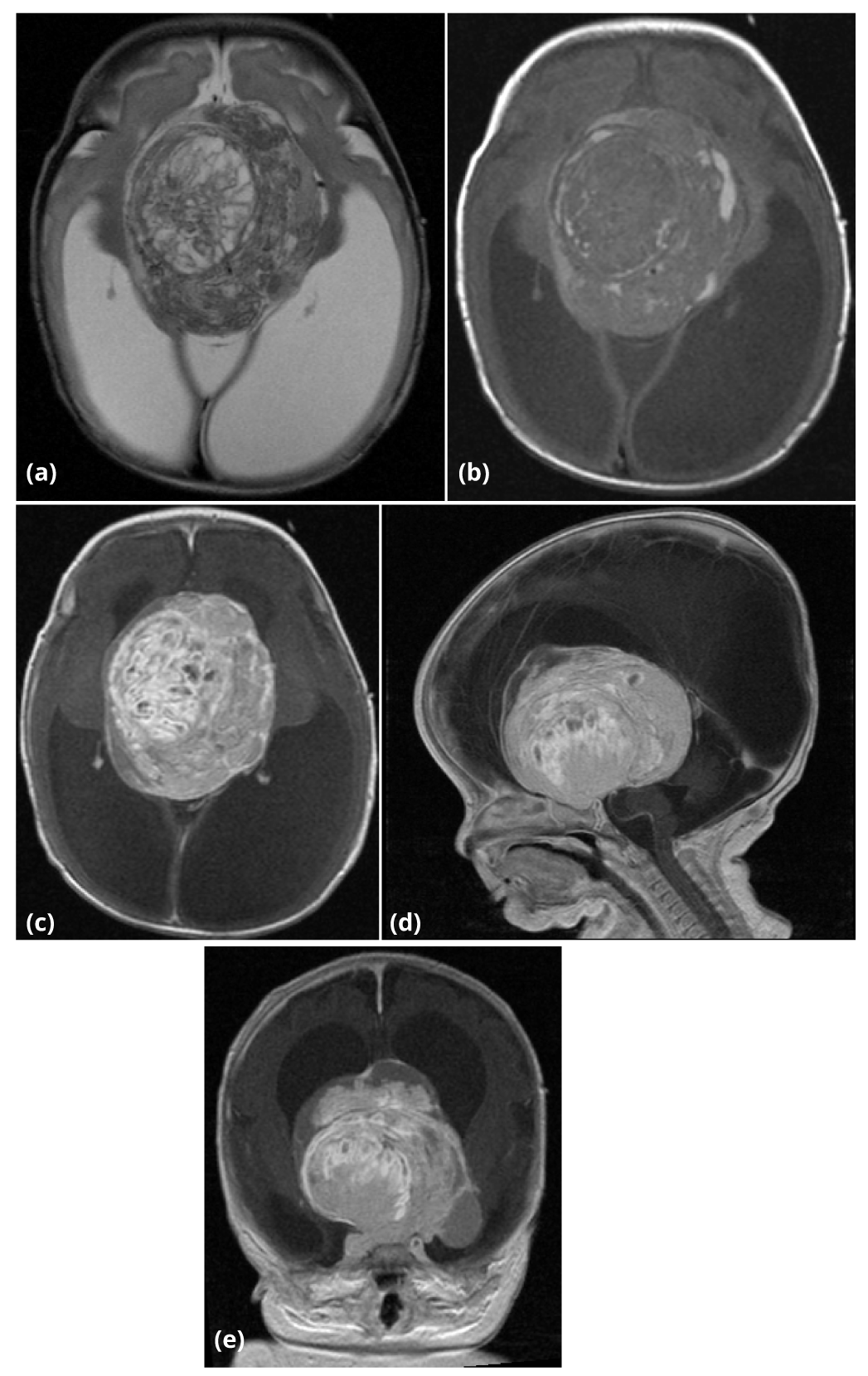
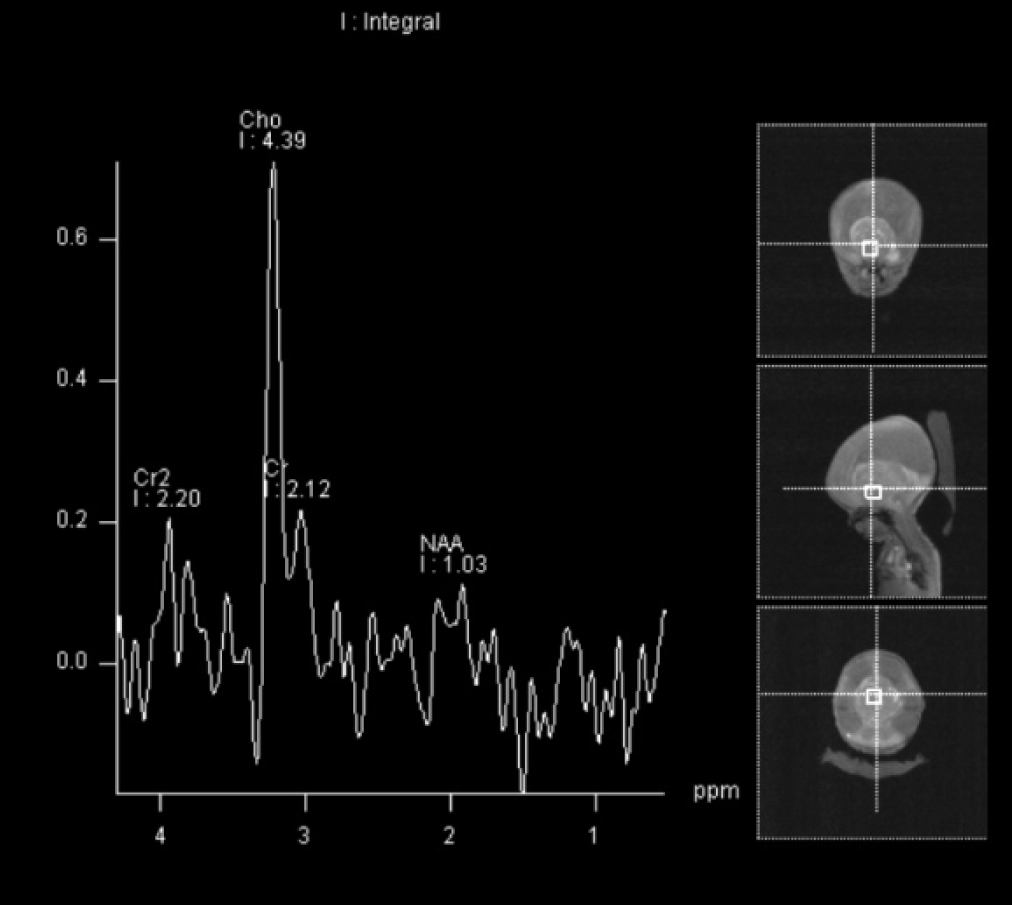
Concurrent screening of the spine was negative for drop metastases. The baby was taken to the operating room on day 4 of life for tumor biopsy and placement of a ventriculoperitoneal shunt. The biopsy demonstrated a craniopharyngioma.
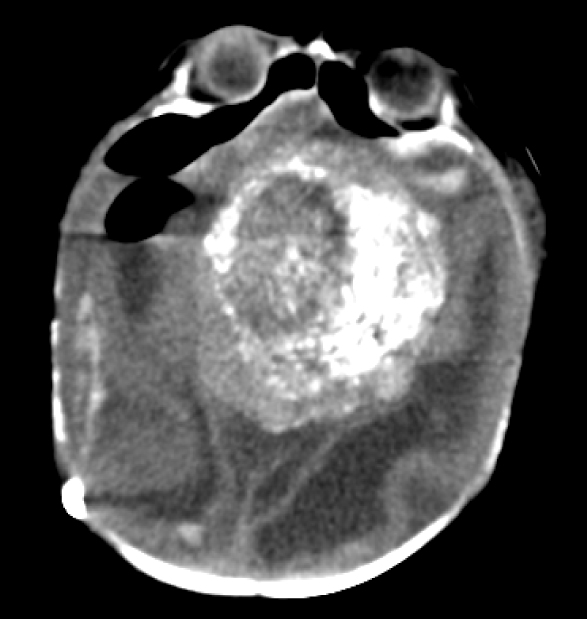
A noncontrasted head CT was obtained after the biopsy. Heterogeneous calcifications within the suprasellar mass that are classically seen in craniopharyngiomas were better appreciated on CT compared to the prior MRI (Figure 5).
At 5 weeks of age, the baby returned to the operating room for attempted excision of the craniopharyngioma due to poor feeding and failure to thrive. However, the patient became hypotensive secondary to uncontrollable bleeding in the resection bed and suffered intra-operative cardiac arrest despite prolonged attempts at resuscitation.
DiscussionTop
Congenital tumors are defined as neoplasms diagnosed prenatally and up to 60 days of age. Congenital intracranial tumors are very rare and only account for 0.5-1.9% of all congenital tumors [1, 3]. Fortunately, as obstetric ultrasound screening has become more commonplace and image quality has improved, early detection of these lesions has increased.
The addition of fetal MRI has allotted radiologists the ability to better characterize these tumors in utero. The early detection can impact decision making regarding termination of the pregnancy, alter the birth plan, guide neonatal critical care, and prompt the appropriate genetic testing. Early successful surgical intervention may also afford the patient lower morbidity post-operatively. An accurate imaging diagnosis can also greatly impact the prognostic data and anticipatory guidance provided to the parents. Based on the fetal MRI, guidance may be provided for a needle biopsy, if termination of the pregnancy is a consideration. Otherwise, prenatal biopsy or immediate postnatal open biopsy has no significant role, as the overall survival does not depend on the histology [1].
A postnatal brain in MRI is usually obtained in the first week of life. The postnatal imaging can be performed with gadolinium contrast and MR spectroscopy, which can provide important additional information about the nature of the lesion, such as high-grade versus low-grade features. The postnatal imaging will also demonstrate how fast the mass is growing and determine if secondary complications have progressed. A MRI with contrast can also help differentiate a mass from a benign vascular malformation or dural arteriovenous malformation, which is often difficult to exclude on ultrasound. If a vascular malformation is present on imaging, then the management course for the patient would drastically change from surgical resection to endovascular embolization.
Of the congenital brain tumors, teratomas are the most common and account for 42% in one review article by Isaacs [1]. The other congenital brain tumors that have been described in the literature include astrocytoma, craniopharyngioma, primitive neuroectodermal tumor, choroid plexus papilloma, meningeal tumors, and ependymoma in order of decreasing frequency [1]. Intriguingly, brain tumors detected in the perinatal period have a supratentorial predilection, unlike the infratentorial location typically seen in later childhood [4]. No matter the histology of the intracranial tumor, macrocephaly, intracranial mass, hydrocephalus, and stillbirth are the most common presentations [1]. Unfortunately, the overall survival rate for congenital brain tumor cases is only 28% [1].
Compared to teratomas, congenital craniopharyngiomas only make up 11% of congenital brain tumor cases. Therefore based upon statistics alone, it is not surprising that the leading diagnosis in the presented case was initially teratoma. Congenital teratomas are typically large intracranial, partially cystic masses that can cause obstructive hydrocephalus or completely replace the normal intracranial tissues. Although teratomas usually arise from the cerebral hemispheres, an intraventricular location within the third and lateral ventricles has also been described in 8% of cases [1]. Despite its benign histology, congenital teratomas have the worst survival rate of all congenital brain tumors. Isaacs’ review showed 41% of the congenital teratoma cases had intrauterine demise and another 41% died within the first week of life. The patient presented in this case report demonstrated some features prenatally that can be seen in congenital teratomas, but postnatal imaging disfavored teratoma and ultimately tissue sampling was required to make the final diagnosis.
Craniopharyngiomas are relatively benign epithelial tumors that are classified WHO grade I and are typically the admantinomatous subtype in pediatric cases [4]. The tumor is thought to arise from remnants of Rathke’s pouch in the suprasellar region and can extend into the hypothalamus and third ventricle [4]. There is a bimodal age distribution in children age 5 to 14 and adults over 50-years-old [5]. Perinatal craniopharyngiomas are extremely rare.
Craniopharyngiomas are mixed solid and cystic lesions often with speckled calcifications. The cystic components can potentially rupture during a vaginal delivery or intra-operatively leading to lethal meningitis [5]. Given the macrocephaly typically associated with craniopharyngiomas and the potential risk of cyst rupture, delivery by cesarean section is usually performed [5].
On obstetric ultrasound in the third trimester, the craniopharyngioma appears as an echogenic midline mass. The sonogram should be carefully evaluated for intra-lesional calcifications, which narrows the differential diagnosis in 60% of cases [5]. Hydrocephalus and increased head circumference may be present depending upon the size of the lesion [2]. Additionally, utilization of power Doppler can potentially demonstrate intra-lesional blood flow or displacement of the intracranial vessels [2]. Polyhydramnios may also be present in patients where the mass causes difficulty swallowing [3]. The differential diagnosis based upon the sonographic findings includes congenital teratoma, astrocytoma, lipoma of the corpus callosum, hypothalamic glioma, and intracranial hemorrhage [2].
Performance of fetal MRI can be vital in narrowing the differential diagnosis. Confirmation of a prenatal diagnosis may provide important information to an obstetrician considering termination of the pregnancy. Postnatal treatment plans can also be discussed prior to the birth. In the event the patient is critically ill in the postnatal period and unable to undergo further imaging safely, the information obtained from the fetal MRI can become even more essential for treatment planning and therefore is a key element in the workup of a congenital brain tumor [4].
On MRI, the signal intensity of the cystic components in a craniopharyngioma will depend upon the degree of proteinaceous contents [3]. If gadolinium contrast is administered in a postnatal MRI, the solid components of the mass will avidly enhance [3]. The classic calcifications of a craniopharyngioma are often better appreciated on CT compared to MRI.
The prognosis of congenital craniopharyngiomas is poor with a 23% survival rate [1]. For comparison, the 10 year survival rate in older children and adults diagnosed with craniopharyngioma is over 90% [6]. The poorer outcomes in perinatal cases are typically related to tumor size and extension into vital structures leading to subtotal resection and complications from severe endocrine dysfunction [1, 6]. Also, the larger the size of the tumor, the greater the chance of adjacent brain parenchymal destruction [2]. The larger tumors are highly associated with intrauterine death, poor survival and poor surgical outcome [3, 4]. Additionally, radiotherapy is contraindicated in neonates thereby limiting treatment options [4]. If complete resection is attained, a 7% risk of recurrence remains [2]. So far 11 cases were reported including our cases (Table 1).
| Report | Live birth | Surgery | Post-surgical survival |
| Kostadinov S et al., [3] | 7 | 5 | 80% |
| Till date including our cases | 8 | 6 | 67% |
There are significant morbidities in survivors after successful resection of a congenital craniopharyngioma. Due to the suprasellar location, these patients are at high risk for atrophy of the optic chiasm and nerves, which can lead to blindness [4]. There is associated hypofunction of the pituitary and diabetes insipidus post-operatively, which requires permanent hormone replacement therapy [4]. Furthermore, the electrolyte disturbances related to the panhypopituitarism can complicate the patient’s intra- and post-operative course [1]. Given the potentially extensive resection, survivors are at risk for delayed psychomotor development and other psychological disorders as well [4]. Nonetheless, as neurosurgical techniques continue to advance, the survival rate continues to improve. Overall, the survival of pediatric craniopharyngiomas is 92% at 5 year and 87% at 10 year [7].
ConclusionTop
Teratomas are the most common intracranial neoplasm in the perinatal period (approximately 42% of cases). However, there is a differential diagnosis including craniopharyngioma, astrocytoma, lipoma of the corpus callosum, hypothalamic glioma, and intracranial hemorrhage. Congenital intracranial neoplasms are very rare and account for 0.5-1.9% of intracranial tumors. Within this rare category, congenital craniopharyngiomas makes up approximately 11%. Intratumoral calcifications are present in 60% of congenital craniopharyngiomas while only 18% of intracranial teratomas contain calcifications. Craniopharyngiomas diagnosed in the perinatal period carry a worse prognosis than those diagnosed in later childhood or adulthood. Definitive diagnosis typically requires a tissue biopsy and surgical resection is often difficult given the suprasellar location. Unfortunately, the patient frequently suffers from blindness and central endocrine dysfunction post-operatively. Intracranial neoplasms are typically detected on third trimester obstetric ultrasound. Further evaluation with fetal MRI is recommended to better characterize the lesion and secondary complications, such as obstructive hydrocephalus. A postnatal brain MRI is also ideally obtained in the first week of life to evaluate interval change, assess enhancement pattern, perform MR spectroscopy, and provide additional information for surgical planning.
Conflicts of interest
The authors declare no conflicts of interest.
ReferencesTop
[1]Isaacs H Jr. Fetal brain tumors: a review of 154 cases. Am J Perinatol. 2009; 26(6):453–466.Article Pubmed
[2]Sosa-Olavarria A, Diaz-Guerrero L, Reigoza A, Bermúdez A, Murillo M. Fetal craniopharyngioma: early prenatal diagnosis. J Ultrasound Med. 2001; 20(7):803–806.Article Pubmed
[3]Kostadinov S, Hanley CL, Lertsburapa T, O’Brien B, He M. Fetal craniopharyngioma: management, postmortem diagnosis, and literature review of an intracranial tumor detected in utero. Pediatr Dev Pathol. 2014; 17(5):409–412.Article Pubmed
[4]Jurkiewicz E, Bekiesińska-Figatowska M, Duczkowski M, Grajkowska W, Roszkowski M, et al. Antenatal diagnosis of the congenital craniopharyngioma. Pol J Radiol. 2010; 75(1):98–102.Pubmed
[5]Joo JG, Rigo J Jr, Sápi Z, Timar B. Foetal craniopharyngioma diagnosed by prenatal ultrasonography and confirmed by histopathological examination. Prenat Diagn. 2009; 29(2):160–163.Article Pubmed
[6]Hwang SW, Su JM, Jea A. Diagnosis and management of brain and spinal cord tumors in the neonate. Semin Fetal Neonatal Med. 2012; 17(4):202–206.Article Pubmed
[7]Pan J, Qi S, Liu Y, Lu Y, Peng J, et al. Growth patterns of craniopharyngiomas: clinical analysis of 226 patients. J Neurosurg Pediatr. 2016; 17(4):418–433.Article Pubmed
 Copyright
© 2012-2025 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2025 NobleResearch Group. All Rights Reserved